
Philosophy of Chemistry
Chemistry is the study of the structure and transformation of matter. When Aristotle wrote the first systematic treatises on chemistry in the 4th century BCE, his conceptual grasp of the nature of matter was tailored to accommodate a relatively simple range of observable phenomena. In the 21st century, chemistry has become the largest scientific discipline, producing over half a million publications a year ranging from direct empirical investigations to substantial theoretical work. However, the specialized interest in the conceptual issues arising in chemistry, hereafter Philosophy of Chemistry, is a relatively recent addition to philosophy of science.
Philosophy of chemistry has two major parts. In the first, conceptual issues arising within chemistry are carefully articulated and analyzed. Such questions which are internal to chemistry include the nature of substance, atomism, the chemical bond, and synthesis. In the second, traditional topics in philosophy of science such as realism, reduction, explanation, confirmation, and modeling are taken up within the context of chemistry.
- 1. Substances, Elements, and Chemical Combination
- 2. Atomism
- 3. The Chemical Revolution
- 4. Structure in Chemistry
- 5. Mechanism and Synthesis
- 6. Chemical Reduction
- 7. Modeling and Chemical Explanation
- Bibliography
- Academic Tools
- Other Internet Resources
- Related Entries
1. Substances, Elements, and Chemical Combination
Our contemporary understanding of chemical substances is elemental and atomic: All substances are composed of atoms of elements such as hydrogen and oxygen. These atoms are the building blocks of the microstructures of compounds and hence are the fundamental units of chemical analysis. However, the reality of chemical atoms was controversial until the beginning of the 20th century and the phrase “fundamental building blocks” has always required careful interpretation. So even today, the claim that all substances are composed of elements does not give us sufficient guidance about the ontological status of elements and how the elements are to be individuated.
In this section, we will begin with the issue of elements. Historically, chemists have offered two answers to the question “What is it for something to be an element?”
- An element is a substance which can exist in the isolated state and which cannot be further analyzed (hereafter the end of analysis thesis).
- An element is a substance which is a component of a composite substance (hereafter the actual components thesis).
These two theses describe elements in different ways. In the first, elements are explicitly identified by a procedure. Elements are simply the ingredients in a mixture that can be separated no further. The second conception is more theoretical, positing elements as constituents of composite bodies. In the pre-modern Aristotelian system, the end of analysis thesis was the favored option. Aristotle believed that elements were the building blocks of chemical substances, only potentially present in these substances. The modern conception of elements asserts that they are actual components, although, as we will see, aspects of the end of analysis thesis linger. This section will explain the conceptual background behind chemistry’s progression from one conception to the other. Along the way, we will discuss the persistence of elements in chemical combination, the connection between element individuation and classification, and criteria for determining pure substances.
1.1 Aristotle’s Chemistry
The earliest conceptual analyses concerning matter and its transformations come in the Aristotelian tradition. As in modern chemistry, the focus of Aristotle’s theories was the nature of substances and their transformations. He offered the first systematic treatises of chemical theory in On Generation and Corruption (De Generatione et Corruptione), Meteorology, and parts of Physics and On the Heavens (De Caelo).
Aristotle recognized that most ordinary, material things are composed of multiple substances, although he thought that some of them could be composed of a single, pure substance. Thus, he needed to give a criterion of purity that would individuate a single substance. His criterion was that pure substances are homoeomerous: they are composed of like parts at every level. “[I]f combination has taken place, the compound must be uniform—any part of such a compound is the same as the whole, just as any part of water is water” (De Generatione et Corruptione, henceforth DG, I.10, 328a10ff).[1] So when we encounter diamond in rock, oil in water, or smoke in air, Aristotelian chemistry tells us that there is more than one substance present.
Like some of his predecessors, Aristotle held that the elements Fire, Water, Air, and Earth were the building blocks of all substances. But unlike his predecessors, Aristotle established this list from fundamental principles. He argued that “it is impossible for the same thing to be hot and cold, or moist and dry … Fire is hot and dry, whereas Air is hot and moist …; and Water is cold and moist, while Earth is cold and dry” (DG II.3, 330a30–330b5). Aristotle supposed hot and moist to be maximal degrees of heat and humidity, and cold and dry to be minimal degrees. Non-elemental substances are characterized by intermediate degrees of the primary qualities of warmth and humidity.
Aristotle used this elemental theory to account for many properties of substances. For example he distinguished between liquids and solids by noting the different properties imposed by two characteristic properties of elements, moist and dry. “[M]oist is that which, being readily adaptable in shape, is not determinable by any limit of its own; while dry is that which is readily determinable by its own limit, but not readily adaptable in shape” (DG II.2, 329b30f.). Solid bodies have a shape and volume of their own, liquids only have a volume of their own. He further distinguished liquids from gases, which don’t even have their own volume. He reasoned that while water and air are both fluid because they are moist, cold renders water liquid and hot makes air gas. On the other hand, dry together with cold makes earth solid, but together with hot we get fire.
Chemistry focuses on more than just the building blocks of substances: It attempts to account for the transformations that change substances into other kinds of substances. Aristotle also contributed the first important analyses of this process, distinguishing between transmutation, where one substance overwhelms and eliminates another and proper mixing. The former is closest to what we would now call change of phase and the latter to what we would now call chemical combination.
Aristotle thought that proper mixing could occur when substances of comparable amounts are brought together to yield other substances called ‘compounds.’[2] Accordingly, the substances we typically encounter are compounds, and all compounds have the feature that there are some ingredients from which they could be made.
What happens to the original ingredients when they are mixed together to form a compound? Like modern chemists, Aristotle argued that the original ingredients can, at least in principle, be obtained by further transformations. He presumably knew that salt and water can be obtained from sea water and metals can be obtained from alloys. But he explains this with a conceptual argument, not a detailed list of observations.
Aristotle first argues that heterogeneous mixtures can be decomposed:
Observation shows that even mixed bodies are often divisible into homoeomerous parts; examples are flesh, bone, wood, and stone. Since then the composite cannot be an element, not every homoeomerous body can be an element; only, as we said before, that which is not divisible into bodies different in form (De caelo, III.4, 302b15–20).
He then goes on to offer an explicit definition of the concept of an element in terms of simple bodies, specifically mentioning recovery in analysis.
An element, we take it, is a body into which other bodies may be analyzed, present in them potentially or in actuality (which of these is still disputable), and not itself divisible into bodies different in form. That, or something like it, is what all men in every case mean by element (De caelo, III.3, 302a15ff).
The notion of simplicity implicit here is introduced late in DG where in book II Aristotle claims that “All the compound bodies … are composed of all the simple bodies” (334b31). But if all simple bodies (elements) are present in all compounds, how are the various compounds distinguished? With an eye to more recent chemistry, it is natural to think that the differing degrees of the primary qualities of warmth and humidity that characterize different substances arise from mixing different proportions of the elements. Perhaps Aristotle makes a fleeting reference to this idea when he expresses the uniformity of a product of mixing by saying that “the part exhibit[s] the same ratio between its constituents as the whole” (DG I.10, 328a8–9 and again at DG II.7, 334b15).
But what does “proportions of the elements” mean? The contemporary laws of constant and multiple proportions deal with a concept of elemental proportions understood on the basis of the concept of mass. No such concept was available to Aristotle. The extant texts give little indication of how Aristotle might have understood the idea of elemental proportions, and we have to resort to speculation (Needham 2009).
Regardless of how he understood elemental proportions, Aristotle was quite explicit that while recoverable, elements were not actually present in compounds. In DG I.10 he argues that the original ingredients are only potentially, and not actually, present in the resulting compounds of a mixing process.
There are two reasons why in Aristotle’s theory the elements are not actually present in compounds. The first concerns the manner in which mixing occurs. Mixing only occurs because of the primary powers and susceptibilities of substances to affect and be affected by other substances. This implies that all of the original matter is changed when a new compound is formed. Aristotle tells us that compounds are formed when the opposing contraries are neutralized and an intermediate state results:
since there are differences in degree in hot and cold, … [when] both by combining destroy one another’s excesses so that there exist instead a hot which (for a hot) is cold and a cold which (for a cold) is hot; then there will exist … an intermediate. … It is thus, then, … that out of the elements there come-to-be flesh and bones and the like—the hot becoming cold and the cold becoming hot when they have been brought to the mean. For at the mean is neither hot nor cold. The mean, however, is of considerable extent and not indivisible. Similarly, it is in virtue of a mean condition that the dry and the moist and the rest produce flesh and bone and the remaining compounds. (DG II.7, 334b8–30)
The second reason has to do with the homogeneity requirement of pure substances. Aristotle tells us that “if combination has taken place, the compound must be uniform—any part of such a compound is the same as the whole, just as any part of water is water” (DG I.10, 328a10f.). Since the elements are defined in terms of the extremes of warmth and humidity, what has intermediate degrees of these qualities is not an element. Being homogeneous, every part of a compound has the same intermediate degrees of these qualities. Thus, there are no parts with extremal qualities, and hence no elements actually present. His theory of the appearance of new substances therefore implies that the elements are not actually present in compounds.
So we reach an interesting theoretical impasse. Aristotle defined the elements by conditions they exhibit in isolation and argued that all compounds are composed of the elements. However, the properties elements have in isolation are nothing that any part of an actually existing compound could have. So how is it possible to recover the elements?
It is certainly not easy to understand what would induce a compound to dissociate into its elements on Aristotle’s theory, which seems entirely geared to showing how a stable equilibrium results from mixing. The overwhelming kind of mixing process doesn’t seem to be applicable. How, for example, could it explain the separation of salt and water from sea water? But the problem for the advocates of the actual presence of elements is to characterize them in terms of properties exhibited in both isolated and combined states. The general problem of adequately meeting this challenge, either in defense of the potential presence or actual presence view, is the problem of mixture (Cooper 2004; Fine 1995, Wood & Weisberg 2004).
In summary, Aristotle laid the philosophical groundwork for all subsequent discussions of elements, pure substances, and chemical combination. He asserted that all pure substances were homoeomerous and composed of the elements air, earth, fire, and water. These elements were not actually present in these substances; rather, the four elements were potentially present. Their potential presence could be revealed by further analysis and transformation.
1.2 Lavoisier’s Elements
Antoine Lavoisier (1743–1794) is often called the father of modern chemistry, and by 1789 he had produced a list of the elements that a modern chemist would recognize. Some items such as hydrogen and oxygen gases were regarded as compounds by Lavoisier, although we now know regard hydrogen and oxygen as elements and their gases as molecules. Other items on his list were remnants of the Aristotelian system which have no place at all in the modern system. For example, fire remained on his list, although in the somewhat altered form of caloric. Air is analyzed into several components: the respirable part called oxygen and the remainder called azote or nitrogen. Four types of earth found a place on his list: lime, magnesia, barytes, and argill. The composition of these earths are “totally unknown, and, until by new discoveries their constituent elements are ascertained, we are certainly authorized to consider them as simple bodies” (1789, p. 157), although Lavoisier goes on to speculate that “all the substances we call earths may be only metallic oxyds” (1789, p. 159).
What is especially important about Lavoisier’s system is his discussion of how the elemental basis of particular compounds is determined. For example, he describes how water can be shown to be a compound of hydrogen and oxygen (1789, pp. 83–96). He writes:
When 16 ounces of alcohol are burnt in an apparatus properly adapted for collecting all the water disengaged during the combustion, we obtain from 17 to 18 ounces of water. As no substance can furnish a product larger than its original bulk, it follows, that something else has united with the alcohol during its combustion; and I have already shown that this must be oxygen, or the base of air. Thus alcohol contains hydrogen, which is one of the elements of water; and the atmospheric air contains oxygen, which is the other element necessary to the composition of water (1789, p. 96).
The metaphysical principle of the conservation of matter—that matter can be neither created nor destroyed in chemical processes—called upon here is at least as old as Aristotle (Weisheipl 1963). What the present passage illustrates is the employment of a criterion of conservation: the preservation of mass. The total mass of the products must come from the mass of the reactants, and if this is not to be found in the easily visible ones, then there must be other, less readily visible reactants.
This principle enabled Lavoisier to put what was essentially Aristotle’s notion of simple substances (302a15ff., quoted in section 1.1) to much more effective experimental use. Directly after rejecting atomic theories, he says “if we apply the term elements, or principles of bodies, to express our idea of the last point which analysis is capable of reaching, we must admit, as elements, all the substances into which we are capable, by any means, to reduce bodies by decomposition” (1789, p. xxiv). In other words, elements are identified as the smallest components of substances that we can produce experimentally. The principle of the conservation of mass provided for a criterion of when a chemical change was a decomposition into simpler substances, which was decisive in disposing of the phlogiston theory. The increase in weight on calcination meant, in the light of this principle, that calcination was not a decomposition, as the phlogiston theorists would have it, but the formation of a more complex compound.
Despite the pragmatic character of this definition, Lavoisier felt free to speculate about the compound nature of the earths, as well as the formation of metal oxides which required the decomposition of oxygen gas. Thus, Lavoisier also developed the notion of an element as a theoretical, last point of analysis concept. While this last point of analysis conception remained an important notion for Lavoisier as it was for Aristotle, his notion was a significant advance over Aristotle’s and provided the basis for further theoretical advance in the 19th century (Hendry 2005).
1.3 Mendeleev’s Periodic Table
Lavoisier’s list of elements was corrected and elaborated with the discovery of many new elements in the 19th century. For example, Humphrey Davy (1778–1829) isolated sodium and potassium by electrolysis, demonstrating that Lavoisier’s earths were actually compounds. In addition, caloric disappeared from the list of accepted elements with the discovery of the first law of thermodynamics in the 1840s. Thus with this changing, but growing, number of elements, chemists increasingly recognized the need for a systematization. Many attempts were made, but an early influential account was given by John Newlands (1837–98) who prepared the first periodic table showing that 62 of the 63 then known elements follow an “octave” rule according to which every eighth element has similar properties.
Later, Lothar Meyer (1830–95) and Dmitrij Mendeleev (1834–1907) independently presented periodic tables covering all 63 elements known in 1869. In 1871, Mendeleev published his periodic table in the form it was subsequently acclaimed. This table was organized on the idea of periodically recurring general features as the elements are followed when sequentially ordered by relative atomic weight. The periodically recurring similarities of chemical behavior provided the basis of organizing elements into groups. He identified 8 such groups across 12 horizontal periods, which, given that he was working with just 63 elements, meant there were several holes.
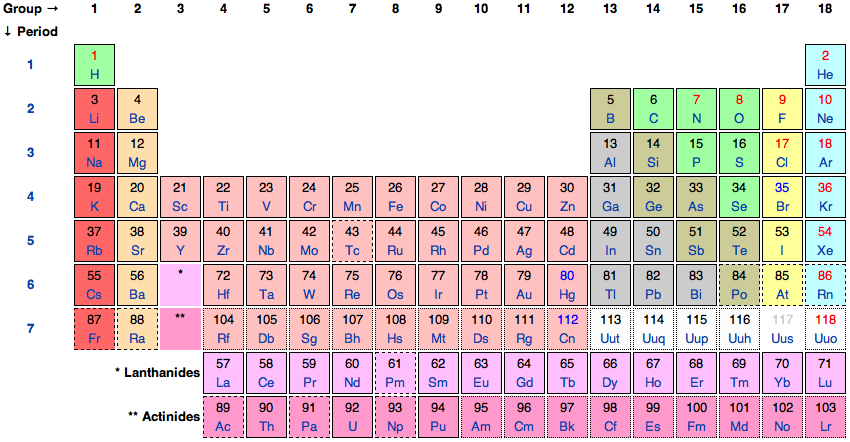
Figure 1. The International Union of Pure and Applied Chemistry’s Periodic Table of the Elements.
The modern Periodic Table depicted in Figure 1 is based on Mendeleev’s table, but now includes 92 naturally occurring elements and some dozen artificial elements (see Scerri 2006). The lightest element, hydrogen, is difficult to place, but is generally placed at the top of the first group. Next comes helium, the lightest of the noble gases, which were not discovered until the end of the 19th century. Then the second period begins with lithium, the first of the group 1 (alkali metal) elements. As we cross the second period, successively heavier elements are first members of other groups until we reach neon, which is a noble gas like helium. Then with the next heaviest element sodium we return to the group 1 alkali metals and begin the third period, and so on.
On the basis of his systematization, Mendeleev was able to correct the values of the atomic weights of certain known elements and also to predict the existence of then unknown elements corresponding to gaps in his Periodic Table. His system first began to seriously attract attention in 1875 when he was able to point out that gallium, the newly discovered element by Lecoq de Boisbaudran (1838–1912), was the same as the element he predicted under the name eka-aluminium, but that its density should be considerably greater than the value Lecoq de Boisbaudran reported. Repeating the measurement proved Mendeleev to be right. The discovery of scandium in 1879 and germanium in 1886 with the properties Mendeleev predicted for what he called “eka-bor” and “eka-silicon” were further triumphs (Scerri 2006).
In addition to providing the systematization of the elements used in modern chemistry, Mendeleev also gave an account of the nature of elements which informs contemporary philosophical understanding. He explicitly distinguished between the end of analysis and actual components conceptions of elements and while he thought that both notions have chemical importance, he relied on the actual components thesis when constructing the Periodic Table. He assumed that the elements remained present in compounds and that the weights of compounds is the sum of the weights of their constituent atoms. He was thus able to use atomic weights as the primary ordering property of the Periodic Table.[3]
Nowadays, chemical nomenclature, including the definition of the element, is regulated by The International Union of Pure and Applied Chemistry (IUPAC). In 1923, IUPAC followed Mendeleev and standardized the individuation criteria for the elements by explicitly endorsing the actual components thesis. Where they differed from Mendeleev is in what property they thought could best individuate the elements. Rather than using atomic weights, they ordered elements according to atomic number, the number of protons and of electrons of neutral elemental atoms, allowing for the occurrence of isotopes with the same atomic number but different atomic weights. They chose to order elements by atomic number because of the growing recognition that electronic structure was the atomic feature responsible for governing how atoms combine and the number of electrons is governed by the requirement of overall electrical neutrality (Kragh 2000).
1.4 Complications for the Periodic System
Mendeleev’s periodic system was briefly called into question with the discovery of the inert gas argon in 1894, which had to be placed outside the existing system after chlorine. But William Ramsay (1852–1916) suspected there might be a whole group of chemically inert substances separating the electronegative halogen group 17 (to which chlorine belongs) and the electropositive alkali metals, and by 1898 he had discovered the other noble gases, which became group 18 on the modern Table.
A more serious challenge arose when the English radiochemist Frederick Soddy (1877–1956) established in 1913 that according to the atomic weight criterion of sameness, positions in the periodic table were occupied by several elements. Adopting Margaret Todd’s (1859–1918) suggestion, Soddy called these elements ‘isotopes,’ meaning “same place.” At the same time, Bohr’s conception of the atom as comprising a positively charged nucleus around which much lighter electrons circulated was gaining acceptance. After some discussion about criteria (van der Vet 1979), delegates to the 1923 IUPAC meeting saved the Periodic Table by decreeing that positions should be correlated with atomic number (number of protons in the nucleus) rather than atomic weight.
Correlating positions in the Periodic Table with whole numbers finally provided a criterion determining whether any gaps remained in the table below the position corresponding to the highest known atomic number. The variation in atomic weight for fixed atomic number was explained in 1932 when James Chadwick (1891–1974) discovered the neutron—a neutral particle occurring alongside the proton in atomic nuclei with approximately the same mass as the proton.
Contemporary philosophical discussion about the nature of the elements begins with the work of Friedrich Paneth (1887–1958), who heavily influenced IUPAC standards and definitions. He was among the first modern chemists to draw an explicit distinction between basic substances — the last point of chemical analysis — and simple substances, the actual components identified in analysis. Paneth argued that the basic substance conception could not serve as the proper basis for explaining the properties of compounds, because basic substances are defined purely by their role in analysis rather than by any intrinsic physical properties, and so lack the resources to ground such explanations. It is the simple substance, the physically characterized component actually present in a compound, that does the explanatory work; the basic substance, being a mere abstraction, can at best name what is present but cannot account for why the compound behaves as it does. Paneth concluded that the chemically important notion of element was therefore “transcendental,” which we interpret to mean “an abstraction over the properties in compounds” (Paneth 1962).
Another strand of the philosophical discussion probes at the contemporary IUPAC definition of elements. According to IUPAC, to be gold is to have atomic number 79, regardless of atomic weight. A logical and intended consequence of this definition is that all isotopes sharing an atomic number count as the same element. Needham (2008) has recently challenged this identification by pointing to chemically salient differences among the isotopes. These differences are best illustrated by the three isotopes of hydrogen: protium, deuterium and tritium. The most striking chemical difference among the isotopes of hydrogen is their different rate of chemical reactions. Because of the sensitivity of biochemical processes to rates of reaction, heavy water (deuterium oxide) is poisonous whereas ordinary water (principally protium oxide) is not. With the development of more sensitive measuring techniques, it has become clear that this is a general phenomenon. Isotopic variation affects the rate of chemical reactions, although these effects are less marked with increasing atomic number. In view of the way chemists understand these differences in behavior, Needham argues that they can reasonably be said to underlie differences in chemical substance. He further argues that the criteria of sameness and difference provided by thermodynamics also suggest that the isotopes should be considered different substances. However, notwithstanding his own view, the places in Mendeleev’s periodic table were determined by atomic number (or nuclear charge), so a concentration on atomic weight would be highly revisionary of chemical classification (Hendry 2006a). It can also be argued that the thermodynamic criteria underlying the view that isotopes are different substances distinguish among substances more finely than is appropriate for chemistry (Hendry 2010c).
1.5 Modern Problems about Mixtures and Compounds
Contemporary theories of chemical combination arose from a fusion of ancient theories of proper mixing and hundreds of years of experimental work, which refined those theories. Yet even by the time that Lavoisier inaugurated modern chemistry, chemists had little in the way of rules or principles that govern how elements combine to form compounds. In this section, we discuss theoretical efforts to provide such criteria.
A first step towards a theory of chemical combination was implicit in Lavoisier’s careful experimental work on water. In his Elements of Chemistry, Lavoisier established the mass proportions of hydrogen and oxygen obtained by the complete reduction of water to its elements. The fact that his results were based on multiple repetitions of this experiment suggests that he assumed compounds like water are always composed of the same elements in the same proportions. This widely shared view about the constant proportions of elements in compounds was first explicitly proclaimed as the law of constant proportions by Joseph Louis Proust (1754–1826) in the first years of the 19th century. Proust did so in response to Claude Louis Berthollet (1748–1822), one of Lavoisier’s colleagues and supporters, who argued that compounds could vary in their elemental composition.
Although primarily a theoretical and conceptual posit, the law of constant proportions became an important tool for chemical analysis. For example, chemists had come to understand that atmospheric air is composed of both nitrogen and oxygen and is not an element. But was air a genuine compound of these elements or some looser mixture of nitrogen and oxygen, that could vary at different times and in different places? The law of constant proportions gave a criterion for distinguishing compounds from genuine mixtures. If air was a compound, then it would always have the same proportion of nitrogen and oxygen and it should further be distinguishable from other compounds of nitrogen and oxygen such as nitrous oxide. If air was not a genuine compound, then it would be an example of a solution, a homogenous mixture of oxygen and nitrogen that could vary in proportions.
Berthollet didn’t accept this rigid distinction between solutions and compounds. He believed that whenever a substance is brought into contact with another, it forms a homogeneous union until further addition of the substance leaves the union in excess. For example, when water and sugar are combined, they initially form a homogenous union. At a certain point, the affinities of water and sugar for one another are saturated, and a second phase of solid sugar will form upon the addition of more sugar. This point of saturation will vary with the pressure and temperature of the solution. Berthollet maintained that just as the amount of sugar in a saturated solution varies with temperature and pressure, the proportions of elements in compounds are sensitive to ambient conditions. Thus, he argued, it is not true that substances are always composed of the same proportions of the element and this undermines the law of constant proportions. But after a lengthy debate, chemists came to accept that the evidence Proust adduced established the law of constant proportions for compounds, which were thereby distinguished from solutions.
Chemists’ attention was largely directed towards the investigation of compounds in the first half of the 19th century, initially with a view to broadening the evidential basis which Proust had provided. For a time, the law of constant proportions seemed a satisfactory criterion of the occurrence of chemical combination. But towards the end of the 19th century, chemists turned their attention to solutions. Their investigation of solutions drew on the new science of thermodynamics, which said that changes of state undergone by substances when they are brought into contact were subjected to its laws governing energy and entropy.
Although thermodynamics provided no sharp distinction between compounds and solutions, it did allow the formulation of a concept for a special case called an ideal solution. An ideal solution forms because its increased stability compared with the separated components is entirely due to the entropy of mixing. This can be understood as a precisification of the idea of a purely mechanical mixture. Compounds, on the other hand, are stabilized by interactions between their constituent components over and above the entropy of mixing. For example, solid sodium chloride is stabilized by the interactions of sodium and chlorine, which react to form sodium chloride. The behavior of real solutions could be contrasted with that of an ideal solution, and it turned out that non-ideality was the rule rather than the exception. Ideality is approached only in certain dilute binary solutions. More often, solutions exhibited behavior which could only be understood in terms of significant chemical interactions between the components, of the sort characteristic of chemical combination.
Long after his death, in the first decades of the 20th century, Berthollet was partially vindicated with the careful characterization of a class of substances that we now call Berthollides. These are compounds whose proportions of elements do not stand in simple relations to one another. Their elemental proportions are not fixed, but vary with temperature and pressure. For example, the mineral wüstite, or ferrous oxide, has an approximate compositional formula of FeO, but typically has somewhat less iron than oxygen.
From a purely macroscopic, thermodynamic perspective, Berthollides can be understood in terms of the minimization of the thermodynamic function called the Gibbs free energy, which accommodates the interplay of energy and entropy as functions of temperature and pressure. Stable substances are ones with minimal Gibbs free energy. On the microscopic scale, the basic microstructure of ferrous oxide is a three-dimensional lattice of ferrous (Fe2+) and oxide (O2-) ions. However, some of the ferrous ions are replaced by holes randomly distributed in the crystal lattice, which generates an increase in entropy compared with a uniform crystal structure. An overall imbalance of electrical charge would be created by the missing ions. But this is countered in ferrous oxide by twice that number of ions from those remaining being converted to ferric (Fe3+) ions. This removal of electrons requires an input of energy, which would make for a less stable structure were it not for the increased entropy afforded by the holes in the crystal structure. The optimal balance between these forces depends on the temperature and pressure, and this is described by the Gibbs free energy function.
Although the law of constant proportions has not survived the discovery of Berthollides and more careful analyses of solutions showed that chemical combination or affinity is not confined to compounds, it gave chemists a principled way of studying how elements combine to form compounds through the 19th century. This account of Berthollides also illustrates the interplay between macroscopic and microscopic theory which is a regular feature of modern chemistry, and which we turn to in the next section.
Chemistry has traditionally distinguished itself from classical physics by its interest in the division of matter into different substances and in chemical combination, the process whereby substances are held together in compounds and solutions. In this section, we have described how chemists came to understand that all substances were composed of the Periodic Table’s elements, and that these elements are actual components of substances. Even with this knowledge, distinguishing pure substances from heterogeneous mixtures and solutions remained a very difficult chemical challenge. And despite chemists’ acceptance of the law of definite proportions as a criterion for substancehood, chemical complexities such as the discovery of the Berthollides muddied the waters.
2. Atomism
Modern chemistry is thoroughly atomistic. All substances are thought to be composed of small particles, or atoms, of the Periodic Table’s elements. Yet until the beginning of the 20th century, much debate surrounded the status of atoms and other microscopic constituents of matter. As with many other issues in philosophy of chemistry, the discussion of atomism begins with Aristotle, who attacked the coherence of the notion and disputed explanations supposedly built on the idea of indivisible constituents of matter capable only of change in respect of position and motion, but not intrinsic qualities. We will discuss Aristotle’s critiques of atomism and Boyle’s response as well as the development of atomism in the 19th and 20th centuries.
2.1 Atomism in Aristotle and Boyle
In Aristotle’s time, atomists held that matter was fundamentally constructed out of atoms. These atoms were indivisible and uniform, of various sizes and shapes, and capable only of change in respect of position and motion, but not intrinsic qualities. Aristotle rejected this doctrine, beginning his critique of it with a simple question: What are atoms made of? Atomists argue that they are all made of uniform matter. But why should uniform matter split into portions not themselves further divisible? What makes atoms different from macroscopic substances which are also uniform, but can be divided into smaller portions? Atomism, he argued, posits a particular size as the final point of division in completely ad hoc fashion, without giving any account of this smallest size or why atoms are this smallest size.
Apart from questions of coherence, Aristotle argued that it was unclear and certainly unwarranted to assume that atoms have or lack particular properties. Why shouldn’t atoms have some degree of warmth and humidity like any observable body? But if they do, why shouldn’t the degree of warmth of a cold atom be susceptible to change by the approach of a warm atom, in contradiction with the postulate that atoms only change their position and motion? On the other hand, if atoms don’t possess warmth and humidity, how can changes in degrees of warmth and humidity between macroscopic substances be explained purely on the basis of change in position and motion?
These and similar considerations led Aristotle to question whether the atomists had a concept of substance at all. There are a large variety of substances discernible in the world—the flesh, blood and bone of animal bodies; the water, rock, sand and vegetable matter by the coast, etc. Atomism apparently makes no provision for accommodating the differing properties of these substances, and their interchangeability, when for example white solid salt and tasteless liquid water are mixed to form brine or bronze statues slowly become green. Aristotle recognized the need to accommodate the creation of new substances with the destruction of old by combination involving the mutual interaction and consequent modification of the primary features of bodies brought into contact. In spite of the weaknesses of his own theory, he displays a grasp of the issue entirely lacking on the part of the atomists. His conception of elements as being few in number and of such a character that all the other substances are compounds derived from them by combination and reducible to them by analysis provided the seeds of chemical theory. Ancient atomism provided none.
Robert Boyle (1627–1691) is often credited with first breaking with ancient and medieval traditions and inaugurating modern chemistry by fusing an experimental approach with mechanical philosophy. Boyle’s chemical theory attempts to explain the diversity of substances, including the elements, in terms of variations of shape and size and mechanical arrangements of what would now be called sub-atomic atoms or corpuscles. Although Boyle’s celebrated experimental work attempted to respond to Aristotelian orthodoxy, his theorizing about atoms had little impact on his experimental work. Chalmers (1993, 2002) documents the total absence of any connection between Boyle’s atomic speculations and his experimental work on the effects of pressure on gases. This analysis applies equally to Boyle’s chemical experiments and chemical theorizing, which was primarily driven by a desire to give a mechanical philosophy of chemical combination (Chalmers 2009, Ch. 6). No less a commentator than Antoine Lavoisier (1743–1794) was quite clear that Boyle’s corpuscular theories did nothing to advance chemistry. As he noted towards the end of the next century, “… if, by the term elements, we mean to express those simple and indivisible atoms of which matter is composed, it is extremely probable we know nothing at all about them” (1789, p. xxiv). Many commentators thus regard Boyle’s empirically-based criticisms of the Aristotelian chemists more important than his own atomic theories. (Banchetti, 2020)
2.2 Atomic Realism in Contemporary Chemistry
Contemporary textbooks typically locate discussions of chemical atomism in the 19th century work of John Dalton (1766–1844). Boyle’s ambitions of reducing elemental minima to structured constellations of mechanical atoms had been abandoned by this time, and Dalton’s theory simply assumes that each element has smallest parts of characteristic size and mass which have the property of being of that elemental kind. Lavoisier’s elements are considered to be collections of such characteristic atoms. Dalton argued that this atomic hypothesis explained the law of constant proportions (see section 1.5).
Dalton’s theory gives expression to the idea of the real presence of elements in compounds. He believed that atoms survive chemical change, which underwrites the claim that elements are actually present in compounds. He assumed that atoms of the same element are alike in their weight. On the assumption that atoms combine with the atoms of other elements in fixed ratios, Dalton claimed to explain why, when elements combine, they do so with fixed proportions between their weights. He also introduced the law of multiple proportions, according to which the elements in distinct compounds of the same elements stand in simple proportions. He argued that this principle was also explained by his atomic theory.
Dalton’s theory divided the chemical community and while he had many supporters, a considerable number of chemists remained unconvinced, adopting an agnostic or explicitly anti-atomistic stance. Part of the reason for this was controversy surrounding the empirical application of Dalton’s atomic theory: How should one estimate atomic weights since atoms were such small quantities of matter? Daltonians argued that although such tiny quantities could not be measured absolutely, they could be measured relative to a reference atom (the natural choice being hydrogen as 1). This still left a problem in setting the ratio between the weights of different atoms in compounds. Dalton assumed that, if only one compound of two elements is known, it should be assumed that they combine in equal proportions. Thus, he understood water, for instance, as though it would have been represented by HO in terms of the formulas that Berzelius was to introduce (Berzelius, 1813). But Dalton’s response to this problem seemed arbitrary. Finding a more natural solution became pressing during the first half of the nineteenth century as more and more elements were being discovered, and the elemental compositions of more and more chemical substances were being determined qualitatively (Duhem 2002; Needham 2004; Chalmers 2005a, 2005b, and 2008).
Dalton’s contemporaries raised other objections as well. Jacob Berzelius (1779–1848) argued that Daltonian atomism provided no explanation of chemical combination, how elements hold together to form compounds (Berzelius, 1815). Since his atoms are intrinsically unchanging, they can suffer no modification of the kind Aristotle thought necessary for combination to occur. Lacking anything like the modern idea of a molecule, Dalton was forced to explain chemical combination in terms of atomic packing. He endowed his atoms with atmospheres of caloric whose mutual repulsion was supposed to explain how atoms pack together efficiently. But few were persuaded by this idea, and what came later to be known as Daltonian atomism abandoned the idea of caloric shells altogether.
The situation was made more complex when chemists realized that elemental composition was not in general sufficient to distinguish substances. Dalton was aware that the same elements sometimes give rise to several compounds; there are several oxides of nitrogen, for example. But given the law of constant proportions, these can be distinguished by specifying the combining proportions, which is what is represented by distinct chemical formulas, for example N2O, NO and N2O3 for different oxides of nitrogen. However, as more organic compounds were isolated and analyzed, it became clear that elemental composition doesn’t uniquely distinguish substances. Distinct compounds with the same elemental composition are called isomers. The term was coined by Berzelius in 1832 when organic compounds with the same composition, but different properties, were first recognized. It was later discovered that isomerism is ubiquitous, and not confined to organic compounds.
Isomers may differ radically in “physical” properties such as melting points and boiling points as well as patterns of chemical reactivity. This is the case with dimethyl ether and ethyl alcohol, which have the compositional formula C2H6O in common, but are represented by two distinct structural formulas: (CH3)2O and C2H5OH. These formulas identify different functional groups, which govern patterns of chemical reactivity. The notion of a structural formula was further elaborated to accommodate other isomers that are even more similar. This was the case with a subgroup of stereoisomers called optical isomers, which are alike in many of their physical properties such as melting points and boiling points and (when first discovered) seemed to be alike in chemical reactivity too. Pasteur famously separated enantiomers (stereoisomers of one another) of tartaric acid by preparing a solution of the sodium ammonium salt and allowing relatively large crystals to form by slow evaporation. Using tweezers, he assembled the crystals into two piles, members of the one having shapes which are mirror images of the shapes of those in the other pile. Optical isomers are so called because they have the distinguishing feature of rotating the plane of plane polarized light in opposite directions, a phenomenon first observed in quartz crystals at the beginning of the 19th century. These isomers are represented by three-dimensional structural formulas which are mirror images of one another as we show in Figure 2.
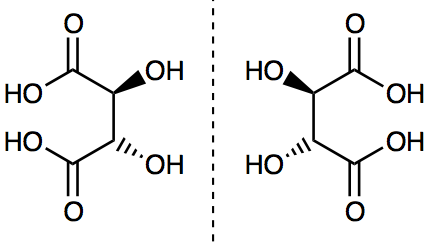
Figure 2. The enantiomers of tartaric acid. D-tartaric acid is on the left and L-tartaric acid is on the right. The dotted vertical line represents a mirror plane. The solid wedges represent bonds coming out of the plane, while the dashed wedges represent bonds going behind the plane. These molecular structures are mirror images of one another.
Although these discoveries are often presented as having been explained by the atomic or molecular hypothesis, skepticism about the status of atomism persisted throughout the 19th century. Late 19th century skeptics such as Ernst Mach, Georg Helm, Wilhelm Ostwald, and Pierre Duhem did not see atomism as an adequate explanation of these phenomena, nor did they believe that there was sufficient evidence to accept the existence of atoms. Instead, they advocated non-atomistic theories of chemical change grounded in thermodynamics (on Helm and Ostwald, see the introduction to Deltete 2000).
Duhem’s objections to atomism are particularly instructive. Despite being represented as a positivist in some literature (e.g. Fox 1971), his objections to atomism in chemistry made no appeal to the unobservability of atoms. Instead, he argued that a molecule was a theoretical impossibility according to 19th century physics, which could say nothing about how atoms can hold together but could give many reasons why they couldn’t be stable entities over reasonable periods of time. He also argued that the notion of valency attributed to atoms to explain their combining power was simply a macroscopic characterization projected into the microscopic level. He showed that chemical formulae could be interpreted without resorting to atoms and the notion of valency could be defined on this basis (Duhem 1892, 1902; for an exposition, see Needham 1996). Atomists failed to meet this challenge, and he criticized them for not saying what the features of their atoms were beyond simply reading into them properties defined on a macroscopic basis (Needham 2004). Duhem did recognize that an atomic theory was developed in the 19th century, the vortex theory (Kragh 2002), but rejected it as inadequate for explaining chemical phenomena.
Evidence for a realm of microscopic particles mounted at the turn of the 20th century. Particularly note-worthy was the careful experimental and theoretical work on Brownian motion, the fluctuation of particles in an emulsion. With the development of kinetic theory it was suspected that this motion was due to invisible particles within the emulsion pushing the visible particles. But it wasn’t until the first decade of the 20th century that Einstein’s theoretical analysis and Perrin’s experimental work gave substance to those suspicions and provided an estimate of Avogadro’s number, which Perrin famously argued was substantially correct because it agreed with determinations made by several other, independent, methods. This was the decisive argument for the existence of microentities which led most of those still skeptical of the atomic hypotheses to change their views (Einstein 1905; Perrin 1913; Nye 1972; Maiocchi 1990).
It is important to appreciate, however, that establishing the existence of atoms in this way left many of the questions raised by the skeptics unanswered. A theory of the nature of atoms which would explain how they can combine to form molecules was yet to be formulated. And it remains to this day an open question whether a purely microscopic theory is available which is adequate to explain the whole range of chemical phenomena. This issue is pursued in Section 6 where we discuss reduction.
3. The Chemical Revolution
As we discussed in Section 1, by the end of the 18th century the modern conception of chemical substances began to take form in Lavoisier’s work. Contemporary-looking lists of elements were being drawn up and also the notion of mass was introduced into chemistry. Despite these advances, chemists continued to develop theories about two substances which we no longer accept: caloric and phlogiston. Lavoisier famously rejected phlogiston, but he accepted caloric. It would be another 60 years until the notion of caloric was finally abandoned with the development of thermodynamics.
3.1 Caloric
In 1761, Joseph Black discovered that heating a body doesn’t always raise its temperature. In particular, he noticed that heating ice at 0°C converts it to liquid at the same temperature. Similarly, there is a latent heat of vaporization which must be supplied for the conversion of liquid water into steam at the boiling point without raising the temperature. It was some time before the modern interpretation of Black’s ground-breaking discovery was fully developed. He had shown that heat must be distinguished from the state of warmth of a body and even from the changes in that state. But it wasn’t until the development of thermodynamics that heating was distinguished as a process from the property or quality of being warm without reference to a transferred substance.
Black himself was apparently wary of engaging in hypothetical explanations of heat phenomena (Fox 1971), but he does suggest an interpretation of the latent heat of fusion of water as a chemical reaction involving the combination of the heat fluid with ice to yield the new substance water. Lavoisier incorporated Black’s conception of latent heat into his caloric theory of heat, understanding latent heat transferred to a body without raising its temperature as caloric fluid bound in chemical combination with that body and not contributing to the body’s degree of warmth or temperature. Lavoisier’s theory thus retains something of Aristotle’s, understanding what we would call a phase change of the same substance as a transformation of one substance into another.
Caloric figures in Lavoisier’s list of elements as the “element of heat or fire” (Lavoisier 1789, p. 175), “becom[ing] fixed in bodies … [and] act[ing] upon them with a repulsive force, from which, or from its accumulation in bodies to a greater or lesser degree, the transformation of solids into fluids, and of fluids to aeriform elasticity, is entirely owing” (1789, p. 183). He goes on to define ‘gas’ as “this aeriform state of bodies produced by a sufficient accumulation of caloric.” Under the list of binary compounds formed with hydrogen, caloric is said to yield hydrogen gas (1789, p. 198). Similarly, under the list of binary compounds formed with phosphorus, caloric yields phosphorus gas (1789, p. 204). The Lavoisian element base of oxygen combines with the Lavoisian element caloric to form the compound oxygen gas. The compound of base of oxygen with a smaller amount of caloric is oxygen liquid (known only in principle to Lavoisier). What we would call the phase change of liquid to gaseous oxygen is thus for him a change of substance. Light also figures in his list of elements, and is said “to have a great affinity with oxygen, … and contributes along with caloric to change it into the state of gas” (1789, p. 185).
3.2 Phlogiston
Another substance concept from roughly the same period is phlogiston, which served as the basis for 18th century theories of processes that came to be called oxidation and reduction. Georg Ernst Stahl (1660–1734) introduced the theory, drawing on older theoretical ideas. Alchemists thought that metals lose the mercury principle under calcination and that when substances are converted to slag, rust, or ash by heating, they lose the sulphur principle. Johann Joackim Becher (1635–82) modified these ideas at the end of the 17th century, arguing that the calcination of metals is a kind of combustion involving the loss of what he called the principle of flammability. Stahl subsequently renamed this principle ‘phlogiston’ and further modified the theory, maintaining that phlogiston could be transferred from one substance to another in chemical reactions, but that it could never be isolated.
For example, metals were thought to be compounds of the metal’s calx and phlogiston, sulphur was thought to be a compound of sulphuric acid and phlogiston, and phosphorus was thought to be a compound of phosphoric acid and phlogiston. Substances such as carbon which left little or no ash after burning were taken to be rich in phlogiston. The preparation of metals from their calxes with the aid of wood charcoal was understood as the transfer of phlogiston from carbon to the metal.
Regarding carbon as a source of phlogiston and no longer merely as a source of warmth was a step forward in understanding chemical reactions (which Ladyman 2011 emphasizes in support of his structural realist interpretation of phlogiston chemistry). The phlogiston theory suggested that reactions could involve the replacement of one part of a substance with another, where previously all reactions were thought to be simple associations or dissociations.
Phlogiston theory was developed further by Henry Cavendish (1731–1810) and Joseph Priestley (1733–1804), who both attempted to better characterize the properties of phlogiston itself. After 1760, phlogiston was commonly identified with what they called ‘inflammable air’ (hydrogen), which they successfully captured by reacting metals with muriatic (hydrochloric) acid. Upon further experimental work on the production and characterizations of these “airs,” Cavendish and Priestley identified what we now call oxygen as ‘dephlogisticated air’ and nitrogen as ‘phlogiston-saturated air.’
As reactants and products came to be routinely weighed, it became clear that metals gain weight when they become a calx. But according to the phlogiston theory, the calx involves the loss of phlogiston. Although the idea that a process involving the loss of a substance could involve the gain of weight seems strange to us, phlogiston theorists were not immediately worried. Some phlogiston theorists proposed explanations based on the ‘levitation’ properties of phlogiston, what Priestly later referred to as phlogiston’s ‘negative weight.’ Another explanation of the phenomenon was that the nearly weightless phlogiston drove out heavy, condensed air from the pores of the calx. The net result was a lighter product. Since the concept of mass did not yet play a central role in chemistry, these explanations were thought to be quite reasonable.
However, by the end of the 1770s, Torbern Olaf Bergman (1735–1784) made a series of careful measurements of the weights of metals and their calxes. He showed that the calcination of metals led to a gain in their weight equal to the weight of oxygen lost by the surrounding air. This ruled out the two explanations given above, but interestingly, he took this in his stride, arguing that, as metals were being transformed into their calxes, they lost weightless phlogiston. This phlogiston combines with the air’s oxygen to form ponderable warmth, which in turn combines with what remains of the metal after loss of phlogiston to form the calx. Lavoisier simplified this explanation by removing the phlogiston from this scheme. This moment is what many call the Chemical Revolution.
4. Structure in Chemistry
Modern chemistry primarily deals with microstructure, not elemental composition. This section will explore the history and consequences of chemistry’s focus on structure. The first half of this section describes chemistry’s transition from a science concerned with elemental composition to a science concerned with structure. The second half will focus on the conceptual puzzles raised by contemporary accounts of bonding and molecular structure.
4.1 Structural Formulas
In the 18th and early 19th centuries, chemical analyses of substances consisted in the decomposition of substances into their elemental components. Careful weighing combined with an application of the law of constant proportions allowed chemists to characterize substances in terms of the mass ratios of their constituent elements, which is what chemists mean by the composition of a compound. During this period, Berzelius developed a notation of compositional formulas for these mass ratios where letters stand for elements and subscripts stand for proportions on a scale which facilitates comparison of different substances. Although these proportions reflect the proportion by weight in grams, the simple numbers are a result of reexpressing gravimetric proportions in terms of chemical equivalents. For example, the formulas ‘H2O’ and ‘H2S’ say that there is just as much oxygen in combination with hydrogen in water as there is sulphur in combination with hydrogen in hydrogen sulphide. However, when measured in weight, ‘H2O’ corresponds to combining proportions of 8 grams of oxygen to 1 gram of hydrogen and ‘H2S’ corresponds to 16 grams of sulphur to 1 of hydrogen in weight.
By the first decades of the 19th century, the nascent sub-discipline of organic chemistry began identifying and synthesizing ever increasing numbers of compounds (Klein 2003). As indicated in section 2.2, it was during this period that the phenomenon of isomerism was recognized, and structural formulas were introduced to distinguish substances with the same compositional formula that differ in their macroscopic properties. Although some chemists thought structural formulas could be understood on a macroscopic basis, others sought to interpret them as representations of microscopic entities called molecules, corresponding to the smallest unit of a compound as an atom was held to be the smallest unit of an element.
In the first half of the nineteenth century there was no general agreement about how the notion of molecular structure could be deployed in understanding isomerism. But during the second half of the century, consensus built around the structural theories of August Kekulé (1829–1896). Kekulé noted that carbon tended to combine with univalent elements in a 1:4 ratio. He argued that this was because each carbon atom could form bonds to four other atoms, even other carbon atoms (1858 [1963], 127). In later papers, Kekulé dealt with apparent exceptions to carbon’s valency of four by introducing the concept of double bonds between carbon atoms. He extended his treatment to aromatic compounds, producing the famous hexagonal structure for benzene (see Rocke 2010), although this was to create a lasting problem for the universality of carbon’s valency of 4 (Brush 1999a, 1999b).
Kekulé’s ideas about bonding between atoms were important steps toward understanding isomerism. Yet his presentations of structure theory lacked a clear system of diagrammatic representation so most modern systems of structural representation originate with Alexander Crum Brown’s (1838–1932) paper about isomerism among organic acids (1864 [1865]). Here, structure was shown as linkages between atoms (see Figure 3).
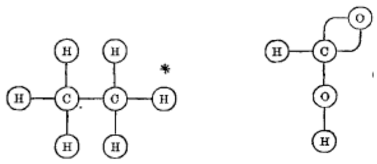
Figure 3. Depictions of ethane and formic acid in Crum Brown’s graphic notation. (1864 [1865], 232)
Edward Frankland (1825–1899) simplified and popularized Crum Brown’s notation in successive editions of his Lecture Notes for Chemical Students (Russell 1971; Ritter 2001). Frankland was also the first to introduce the term ‘bond’ for the linkages between atoms (Ramberg 2003).
The next step in the development of structural theory came when James Dewar (1842–1943) and August Hofmann (1818–1892) developed physical models corresponding closely to Crum Brown’s formulae (Meinel 2004). Dewar’s molecules were built from carbon atoms represented by black discs placed at the centre of pairs of copper bands. In Hofmann’s models, atoms were colored billiard balls (black for carbon, white for hydrogen, red for oxygen etc.) linked by bonds. Even though they were realized by concrete three-dimensional structures of croquet balls and connecting arms, the three-dimensionality of these models was artificial. The medium itself forced the representations of atoms to be spread out in space. But did this correspond to chemical reality?
Kekulé, Crum Brown, and Frankland were extremely cautious when answering this question. Kekulé distinguished between the apparent atomic arrangement which could be deduced from chemical properties, which he called “chemical structure,” and the true spatial arrangement of the atoms (Rocke 1984, 2010). Crum Brown made a similar distinction, cautioning that in his graphical formulae he did not “mean to indicate the physical, but merely the chemical position of the atoms” (Crum Brown, 1864, 232). Frankland noted that “It must carefully be borne in mind that these graphic formulae are intended to represent neither the shape of the molecules, nor the relative position of the constituent atoms” (Biggs et al. 1976, 59).
One way to interpret these comments is that they reflect a kind of anti-realism: Structural formulae are merely theoretical tools for summarizing a compound’s chemical behavior. Or perhaps they are simply agnostic, avoiding definite commitment to a microscopic realm about which little can be said. However, other comments suggest a realist interpretation, but one in which structural formulae represent only the topological structure of the spatial arrangement:
The lines connecting the different atoms of a compound, and which might with equal propriety be drawn in any other direction, provided they connected together the same elements, serve only to show the definite disposal of the bonds: thus the formula for nitric acid indicates that two of the three constituent atoms of oxygen are combined with nitrogen alone, whilst the third oxygen atom is combined both with nitrogen and hydrogen (Frankland, quoted in Biggs et al. 1976, 59; also see Hendry 2010b).
The move towards a fully spatial interpretation was advanced by the simultaneous postulation in 1874 of a tetrahedral structure for the orientation of carbon’s four bonds by Jacobus van ’t Hoff (1852–1911) and Joseph Achille Le Bel (1847–1930) to account for optical isomerism (see Figure 4 and section 2.2). When carbon atoms are bonded to four different constituents, they cannot be superimposed on their mirror images, just as your left and right hands cannot be. This gives rise to two possible configurations of chiral molecules, thus providing for a distinction between distinct substances whose physical and chemical properties are the same except for their ability to rotate plane polarized light in different directions.
van ’t Hoff and Le Bel provided no account of the mechanism by which chiral molecules affect the rotation of plane polarized light (Needham 2004). But by the end of the century, spatial structure was being put to use in explaining the aspects of the reactivity and stability of organic compounds with Viktor Meyer’s (1848–1897) conception of steric hindrance and Adolf von Baeyer’s (1835–1917) conception of internal molecular strain (Ramberg 2003).
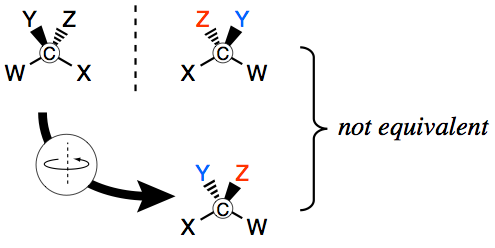
Figure 4. A schematic representation of the tetrahedral arrangement of substituents around the carbon atom. Compare the positions of substituents Y and Z.
Given that these theories were intrinsically spatial, traditional questions about chemical combination and valency took a new direction: What is it that holds the atoms together in a particular spatial arrangement? The answer, of course, is the chemical bond.
4.2 The Chemical Bond
As structural theory gained widespread acceptance at the end of the 19th century, chemists began focusing their attention on what connects the atoms together, constraining the spatial relationships between these atoms. In other words, they began investigating the chemical bond. Modern theoretical accounts of chemical bonding are quantum mechanical, but even contemporary conceptions of bonds owe a huge amount to the classical conception of bonds developed by G.N. Lewis at the very beginning of the 20th century.
The Classical Chemical Bond
G.N. Lewis (1875–1946) was responsible for the first influential theory of the chemical bond (Lewis 1916, 1923; Kossel, 1916; see Kohler 1971, 1975 for background). His theory said that chemical bonds are pairs of electrons shared between atoms. Lewis also distinguished between what came to be called ionic and covalent compounds, which has proved to be remarkably resilient in modern chemistry.
Ionic compounds are composed of electrically charged ions, usually arranged in a neutral crystal lattice. Neutrality is achieved when the positively charged ions (cations) are of exactly the right number to balance the negatively charged ions (anions). Crystals of common salt, for example, comprise as many sodium cations (Na+) as there are chlorine anions (Cl−). Compared to the isolated atoms, the sodium cation has lost an electron and the chlorine anion has gained an electron.
Covalent compounds, on the other hand, are either individual molecules or indefinitely repeating structures. In either case, Lewis thought that they are formed from atoms bound together by shared pairs of electrons. Hydrogen gas is said to consist of molecules composed of two hydrogen atoms held together by a single, covalent bond; oxygen gas, of molecules composed of two oxygen atoms and a double bond; methane, of molecules composed of four equivalent carbon-hydrogen single bonds, and silicon dioxide (sand) crystals of indefinitely repeating covalently bonded arrays of SiO2 units.
An important part of Lewis’ account of molecular structure concerns directionality of bonding. In ionic compounds, bonding is electrostatic and therefore radially symmetrical. Hence an individual ion bears no special relationship to any one of its neighbors. On the other hand, in covalent or non-polar bonding, bonds have a definite direction; they are located between atomic centers.
The nature of the covalent bond has been the subject of considerable discussion in the recent philosophy of chemistry literature (Berson 2008; Hendry 2008; Weisberg 2008; Seifert 2023). While the chemical bond plays a central role in chemical predictions, interventions, and explanations, it is a difficult concept to define precisely. Fundamental disagreements exist between classical and quantum mechanical conceptions of the chemical bond, and even between different quantum mechanical models. Once one moves beyond introductory textbooks to advanced treatments, one finds many theoretical approaches to bonding, but few if any definitions or direct characterizations of the bond itself. While some might attribute this lack of definitional clarity to common background knowledge shared among all chemists, we believe this reflects uncertainty or maybe even ambivalence about the status of the chemical bond itself.
4.3 The Structural Conception of Bonding and its Challenges
The new philosophical literature about the chemical bond begins with the structural conception of chemical bonding (Hendry 2008). On the structural conception, chemical bonds are sub-molecular, material parts of molecules, which are localized between individual atomic centers and are responsible for holding the molecule together. This is the notion of the chemical bond that arose at the end of the 19th century, which continues to inform the practice of synthetic and analytical chemistry. But is the structural conception of bonding correct? Several distinct challenges have been raised in the philosophical literature.
The first challenge comes from the incompatibility between the ontology of quantum mechanics and the apparent ontology of the chemical bonds. Electrons cannot be distinguished in principle (Identity and Individuality in Quantum Theory) and hence quantum mechanical descriptions of bonds cannot depend on the identity of particular electrons. If we interpret the structural conception of bonding in a Lewis-like fashion, where bonds are composed of specific pairs of electrons donated by particular atoms, we can see that this picture is incompatible with quantum mechanics. A related objection notes that both experimental and theoretical evidence suggest that electrons are delocalized, “smeared out” over whole molecules. Quantum mechanics tells us not to expect pairs of electrons to be localized between bonded atoms. Furthermore, Mulliken argued that pairing was unnecessary for covalent bond formation. Electrons in a hydrogen molecule “are more firmly bound when they have two hydrogen nuclei to run around than when each has only one. The fact that two electrons become paired … seems to be largely incidental” (1931, p. 360). Burrau (1927) gave an exact solution (given the Born-Oppenheimer assumption) to the H2+ molecule involving just one electron, which is often referred to in support of Mulliken’s contention.
Defenders of the structural conception of bonding respond to these challenges by noting that G.N. Lewis’ particular structural account isn’t the only possible one. While bonds on the structural conception must be sub-molecular and directional, they need not be electron pairs. Responding specifically to the challenge from quantum ontology, they argue that bonds should be individuated by the atomic centers they link, not by the electrons. Insofar as electrons participate physically in the bond, they do so not as individuals. All of the electrons are associated with the whole molecule, but portions of the electron density can be localized. To the objection from delocalization, they argue that all the structural account requires is that some part of the total electron density of the molecule is responsible for the features associated with the bond and there need be no assumption that it is localized directly between the atoms as in Lewis’ model (Hendry 2008, 2010b).
A second challenge to the structural conception of bonding comes from computational chemistry, the application of quantum mechanics to make predictions about chemical phenomena. Drawing on the work of quantum chemist Charles Coulson (1910–1974), Weisberg (2008) has argued that the structural conception of chemical bonding is not robust in quantum chemistry. This argument looks to the history of quantum mechanical models of molecular structure. In the earliest quantum mechanical models, something very much like the structural conception of bonding was preserved; electron density was, for the most part, localized between atomic centers and was responsible for holding molecules together. However, these early models made empirical predictions about bond energies and bond lengths that were only in qualitative accord with experiment.
Subsequent models of molecular structure yielded much better agreement with experiment when electron density was “allowed” to leave the area between the atoms and delocalize throughout the molecule. As the models were further improved, bonding came to be seen as a whole-molecule, not sub-molecular, phenomenon. Weisberg argues that such considerations should lead us to reject the structural conception of bonding and replace it with a molecule-wide conception. One possibility is the energetic conception of bonding that says that bonding is the energetic stabilization of molecules. Strictly speaking, according to this view, chemical bonds do not exist; bonding is real, bonds are not (Weisberg 2008; also see Coulson 1952, 1960).
The challenges to the structural view of bonding have engendered several responses in the philosophical and chemical literatures. The first appeals to chemical practice: Chemists engaged in synthetic and analytic activities rely on the structural conception of bonding. There are well over 100,000,000 compounds that have been discovered or synthesized, all of which have been formally characterized. How can this success be explained if a central chemical concept such as the structural conception of the bond does not pick out anything real in nature? Throughout his life, Linus Pauling (1901–1994) defended this view.
Another line of objection comes from Berson (2008), who discusses the significance of very weakly bonded molecules. For example, there are four structural isomers of 2-methylenecyclopentane-1,3-diyl. The most stable of the structures does not correspond to a normal bonding interaction because of an unusually stable singlet state, a state where the electron spins are parallel. Berson suggests that this is a case where “the formation of a bond actually produces a destabilized molecule.” In other words, the energetic conception breaks down because bonding and molecule-wide stabilization come apart.
Finally, the “Atoms in Molecules” program (Bader 1991; see Gillespie and Popelier 2001, Chs. 6 & 7 for an exposition) suggests that we can hold on to the structural conception of the bond understood functionally, but reject Lewis’ ideas about how electrons realize this relationship. Bader, for example, argues that we can define ‘bond paths’ in terms of topological features of the molecule-wide electron density. Such bond paths have physical locations, and generally correspond closely to classical covalent bonds. Moreover they partially vindicate the idea that bonding involves an increase in electron density between atoms: a bond path is an axis of maximal electron density (leaving a bond path in a direction perpendicular to it involves a decrease in electron density). There are also many technical advantages to this approach. Molecule-wide electron density exists within the ontology of quantum mechanics, so no quantum-mechanical model could exclude it. Further, electron density is considerably easier to calculate than other quantum mechanical properties, and it can be measured empirically using X-ray diffraction techniques.
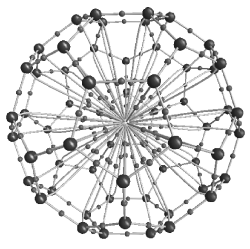
Figure 5. Too many bonds? 60 bond paths from each carbon atom in C60 to a trapped Ar atom in the interior.
Unfortunately, Bader’s approach does not necessarily save the day for the structural conception of the bond. His critics point out that his account is extremely permissive and puts bond paths in places that seem chemically suspect. For example, his account says that when you take the soccer-ball shaped buckminster fullerene molecule (C60) and trap an argon atom inside it, there are 60 bonds between the carbon atoms and the argon atom as depicted in Figure 5 (Cerpa et al. 2008). Most chemists would think this implausible because one of the most basic principles of chemical combination is the fact that argon almost never forms bonds (see Bader 2009 for a response).
A generally acknowledged problem for the delocalized account is the lack of what chemists call transferability. Central to the structural view, as we saw, is the occurrence of functional groups common to different substances. Alcohols, for example, are characterized by having the hydroxyl OH group in common. This is reflected in the strong infra red absorption at 3600cm–1 being taken as a tell-tale sign of the OH group. But ab initio QM treatments just see different problems posed by different numbers of electrons, and fail to reflect that there are parts of a molecular structure, such as an OH group, which are transferable from one molecule to another, and which they may have in common (Woody 2000, 2012).
A further issue is the detailed understanding of the cause of chemical bonding. For many years, the dominant view, based on the Hellman-Feynman theorem, has been that it is essentially an electrostatic attraction between positive nuclei and negative electron clouds (Feynman 1939). But an alternative, originally suggested by Hellman and developed by Rüdenberg, has recently come into prominence. This emphasizes the quantum mechanical analogue of the kinetic energy (Needham 2014). Contemporary accounts may draw on a number of subtle quantum mechanical features. But these details shouldn’t obscure the overriding thermodynamic principle governing the formation of stable compounds by chemical reaction. As Atkins puts it,
Although … the substances involved have dropped to a lower energy, this is not the reason why the reaction takes place. Overall the energy of the Universe remains constant; … All that has happened is that some initially localized energy has dispersed. That is the cause of chemical change: in chemistry as in physics, the driving force of natural change is the chaotic, purposeless, undirected dispersal of energy. (Atkins 1994, p. 112)
The difficulties faced by this and every other model of bonding have led a number of chemists and philosophers to argue for pluralism. Quantum chemist Roald Hoffmann writes “A bond will be a bond by some criteria and not by others … have fun with the concept and spare us the hype” (Hoffmann 2009, Other Internet Resources).
4.4 Molecular Structure and Molecular Shape
While most of the philosophical literature about molecular structure and geometry is about bonding, there are a number of important questions concerning the notion of molecular structure itself. The first issue involves the correct definition of molecular structure. Textbooks typically describe a molecule’s structure as the equilibrium position of its atoms. Water’s structure is thus characterized by 104.5º angles between the hydrogen atoms and the oxygen atom. But this is a problematic notion because molecules are not static entities. Atoms are constantly in motion, moving in ways that we might describe as bending, twisting, rocking, and scissoring. Bader therefore argues that we should think of molecular structure as the topology of bond paths, or the relationships between the atoms that are preserved by continuous transformations (Bader 1991).
A second issue concerning molecular structure is even more fundamental: Do molecules have the kinds of shapes and directional features that structural formulas represent? Given the history we have discussed so far it seems like the answer is obviously yes. Indeed, a number of indirect experimental techniques including x-ray crystallography, spectroscopy, and product analysis provide converging evidence of not only the existence of shape, but specific shapes for specific molecular species.
Despite this, quantum mechanics poses a challenge to the notion of molecular shape. Because of its reliance on the Born-Oppenheimer assumption in quantum mechanical treatments of molecular species, it seems that shape doesn’t arise from quantum mechanics but has to be put in by hand (Woolley 1978; Primas 1981). Sutcliffe and Woolley develop this point further, arguing that the Born-Oppenheimer approximation is not merely a calculational convenience, a simplification that could be relaxed while remaining within the same quantum mechanical framework, but a conceptually distinct assumption that introduces classical geometric structure from outside that framework (Sutcliffe & Woolley 2012). On this view, molecular shape is not a quantum mechanical result at all, but a precondition that must be assumed before the quantum mechanical treatment can even get started. However, recent study of the hydrogen molecule without appeal to the Born-Oppenheimer assumption Mátyus, et al. (2011) shows that the electron density distribution about the nuclei is as expected.
This tension the between the familiar theories of chemical structure and quantum- mechanical accounts of molecules might be resolved in several ways. One might embrace eliminativism about molecular structure: Quantum mechanics is a more fundamental theory, we might argue, and its ontology has no place for molecular structure. Therefore, molecular structure doesn’t exist. No philosopher or chemist that we are aware of has endorsed this option. Another possible response makes a different appeal to the underlying physics. Something must be breaking the wavefunction symmetries and giving atoms locations in molecules. This might be interactions with other molecules or interactions with measuring devices. Thus, molecular shape is partially constituted by interaction and is a relational, not intrinsic property (Ramsey 1997).
A related option is a kind of pragmatism. Hans Primas argues that, strictly speaking, a quantum mechanical description of a molecule has to be a whole-universe description. No matter how we draw the boundaries of interest around some target molecular system, in reality, the system is open and interacting with everything else in the universe. Thus the shape of any particular molecule could be the result of its interactions with anything else in the universe. We only get the paradox of molecules having no shape when we treat systems as closed—say a single methane molecule alone in the universe. It is fine to treat open systems as closed for pragmatic purposes, but we should always understand that this is an idealization. We shouldn’t treat our idealizations, such as open systems being closed, as veridical. Hence there is no incompatibility between quantum mechanics and molecular shape (Primas 1981).
So despite the ubiquity of structural representations of molecules, it turns out that even the notion of molecular shape is not ambiguous. Primas’ approach, which points to the idealization in many quantum mechanical models, is accepted by many chemists. But there is nothing like a consensus in the philosophical literature about how to understand molecular shape.
4.5 Microessentialism: Is Water H2O?
In the final part of this section about structure, we consider a favorite example of philosophers: the thesis that “Water is H2O.” This thesis is often taken to be uncontroversially true and is used as evidence for semantic externalism and for essentialism about natural kinds (Kripke 1980; Putnam 1975, 1990). Since general theses about the theory of reference and semantic externalism are beyond the scope of this article, we focus narrowly on chemical essentialism. Is having a common essential microstructure sufficient to individuate chemical kinds and explain their general features? And if so, is “being H2O” sufficient to individuate water?
The essentialist thesis is often stylized by writing “water = H2O” or “(all and only) water is H2O”. Ignoring the issue of whether the identity makes sense (Needham 2000) and of understanding what the predicates apply to in the second formulation (Needham 2010a), it is not clear that either formulation expresses the kind of thesis that essentialists intend. “H2O” is not a description of any microstructure. Rather, “H2O” is a compositional formula, describing the combining proportions of hydrogen and oxygen to make water.
A reasonable paraphrase of the standard formulation would be “Water is a collection of H2O molecules.” However, although the expression “H2O molecule” describes a particular microentity, it by no means exhausts the kinds of microparticles in water, and says nothing of the microstructure by which they are related in water. Describing the microstructure of water completely involves elaborating the details of this interconnected structure, as well as detailing how they depend on temperature and pressure, and how they change over time (Finney 2004).
Like many other substances, water cannot simply be described as a collection of individual molecules. Here are just a few examples of the complexities of its microstructure: water self-ionizes, which means that hydrogen and hydroxide ions co-exist with H2O molecules in liquid water, continually recombining to form H2O molecules. At the same time, the H2O molecules associate into larger polymeric species. Mentioning these complexities isn’t just pedantic because they are often what give rise to the most striking characteristics of substances. For example, the electrical conductivity of water is due to a mechanism in which a positive charge (hydrogen ion) attaches at one point of a polymeric cluster, inducing a co-ordinated transfer of charge across the cluster, releasing a hydrogen ion at some distant point. The effect is that charge is transferred from one point to another without a transfer of matter to carry it. The hydrogen bonding underlying the formation of clusters is also at the root of many other distinctive properties of water including its high melting and boiling points and its increase in density upon melting. As van Brakel has argued (1986, 2000), water is practically the poster child for such “non-molecular” substances.
Maybe water isn’t simply a collection of H2O molecules, but it certainly has a microstructure and perhaps the essentialist thesis could be recast along the lines of “Water is whatever has its microstructure,” writing in the information that would save this from tautology. But this thesis still endorses the idea that “water” is a predicate characterized by what Putnam calls stereotypical features and exemplified by a paradigm sample identified by an act of ostension. This neglects the importance of macroscopic, yet scientifically important, properties such as boiling points, specific heats, latent heats, and so on, from which much of the microstructure is actually inferred. Indeed, many of the criteria that chemists use to determine the sameness and purity of substances are macroscopic. In fact, international standards for determining the purity of substances like water depend on the careful determination of macroscopic properties such as the triple-point, the temperature and pressure where liquid, gaseous, and solid phases exist simultaneously (Needham 2011). Further, although the idea of a paradigm sample is relevant to the identification of new substances, it is not given by ostension (Needham 2017a).
So is water H2O? In the end, the answer to this question comes down to how one interprets this sentence. Many chemists would be surprised to find out that water wasn’t H2O, but perhaps this is because they read “H2O” as a shorthand (Weisberg 2005) or as a compositional formula in the manner we discussed in the opening of this section. Water is actually characterized by making reference to both its microstructural and macroscopic features, so this can’t on its own provide a justification for microessentialism.
For these reasons, microessentialist claims would need to be grounded in chemical classification and explanation: the systems of nomenclature developed by IUPAC are based entirely on microstructure, as are theoretical explanations of the chemical and spectroscopic behaviour of substances (see Hendry 2016). On the other hand, H2O content fails to track usage of the term “water” by ordinary-language speakers, who seem to have different interests to chemists (Malt 1994). Pluralism is one response to these tensions: Hasok Chang (2012) urges that even within science, water’s identity with H2O should be left open; Julia Bursten (2014) tries to reconcile the special role of microstructure in chemistry with the failure of microessentialism; and Joyce Havstad (2018) argues that chemists’ use of substance concepts is just as messy and disunified as biologists’ use of various species concepts. (See Seifert 2020a, 2020b, and Seifert & Franklin 2024 for further discussion.)
5. Mechanism and Synthesis
Our discussion so far has focused on “static” chemistry: accounts of the nature of matter and its structure. But much of chemistry involves the transformation of matter from one form to another. This section describes the philosophical issues surrounding the synthesis of one substance from another, as well as chemical mechanisms, the explanatory framework chemists use to describe these transformations.
5.1 Mechanistic Explanations in Chemistry
There has been a profusion of discussion in the literatures of philosophy of biology and philosophy of neuroscience about the notion of mechanisms and mechanistic explanations (e.g., Machamer, Darden, & Craver 2000). Yet the production of mechanisms as explanatory schemes finds its original home in chemistry, especially organic chemistry. Chemical mechanisms are used to classify reactions into types, to explain chemical behavior, and to make predictions about novel reactions or reactions taking place in novel circumstances (Weininger 2014).
Goodwin (2012) identifies two notions of chemical mechanism at play in chemistry. The first or thick notion of mechanism is like a motion picture of a chemical reaction. Such a mechanism traces out the positions of all of the electrons and atomic cores of some set of molecules during the course of a reaction, and correlates these positions to the potential energy or free energy of the system. One might think of this as an ideal reaction mechanism, as it would contain all information about the time course of a chemical reaction.
In contrast, the thin notion of a reaction mechanism focuses on a discrete set of steps. In each step, a set of reactive intermediates are generated. These intermediates are quasi-stable molecular species that will ultimately yield the products of the reaction. For example, the much studied biomolecular nucleophilic substitution (SN2) reaction is said to have a single reactive intermediate with the incoming nucleophile and outgoing leaving group both partially bonded to the reactive carbon center (see Figure 6). Such a description of the reaction mechanism is not only abstract in that it leaves out much detail, but it is also highly idealized. Reactions do not take actually place as a series of discrete steps, each of which generates a quasi-stable reaction intermediate.
Figure 6. Thin reaction mechanism for the SN2 reaction.
While most textbook treatments of reaction mechanisms begin by mentioning the thick notion, the details nearly always turn to thin notions of mechanism. At the same time, formal theoretical treatments of reaction mechanisms deal exclusively with the thick notion. Such treatments often attempt to calculate the potential energy function for the reaction from quantum mechanics. Given the importance of the thick notion to formal chemical theorizing, why does the thin notion dominate the practice of chemistry and find expression in textbooks and research articles?
Part of the reason that thin reaction mechanisms are widely used is that determining thick reaction mechanisms is essentially impossible experimentally, and extremely difficult theoretically. But this cannot be the whole story because when necessary, chemists have been able to produce relevant portions of the thick mechanism.
Alternatively, Goodwin (2012, p. 311) has argued that, given the explanatory and predictive goals of chemists, not all of the thick mechanism is needed. In fact, only a characterization of specific structures, the transition state and stable reactive intermediates, are necessary to produce chemical explanations and predictions. Constructing mechanisms as a discrete series of steps between stable and reactive structures allows the chemist:
… to identify or specify which of those standard changes is acting as a “bottleneck” (or in chemical parlance, the “rate determining step”) for the progression of the reaction … make it obvious where alternatives paths are and are not available … [and] to infer something about the structures of the important intermediates in the particular reaction whose mechanism is being proposed (Goodwin 2012, p. 314).
So chemists’ explanatory goals require that specific features of reaction mechanisms can be identified. The rest of the thick mechanism wouldn’t necessarily add any explanatorily relevant detail to the explanation.
5.2 Confirmation of Reaction Mechanisms
Chemists typically do not engage in philosophical discussions about their work. Yet, when discussing the confirmation of reaction mechanisms, it is not uncommon to see mention of philosophical issues surrounding confirmation. So why does the study of reaction mechanisms make chemists more philosophically reflective?
For one thing, almost all studies aimed at elucidating reaction mechanisms rely on indirect techniques. Ideally, elucidating a reaction mechanism would be like doing experiments in biomechanics. Slow motion video could give direct information about the movement of parts and how these movements give rise to the motion of the whole. But we have nothing like a video camera for chemical reactions. Instead, after an experimental determination of the reaction products and possible isolation of stable intermediate species, chemists rely on measurements of reaction rates in differing conditions, spectroscopy, and isotopic labeling, among other techniques. These techniques help eliminate candidate reaction mechanisms, but do not themselves directly suggest new ones. This emphasis on eliminating possibilities has led some chemists to endorse a Popperian, falsificationist analysis of reaction mechanism elucidation (e.g., Carpenter 1984).
Although some chemists have been attracted to a falsificationist analysis, a better analysis of reaction mechanism elucidation is the account of confirmation known as eliminative induction. This account shares falsification’s emphasis on trying to reject hypotheses, but argues that the hypotheses not rejected receive some degree of confirmation. So in the case of reaction mechanisms, we might see eliminative induction as a processes whereby chemists:
- Enumerate reasonable candidates for the reaction mechanism.
- Consider the experimental consequences of these mechanisms, drawing up lists of reaction conditions under which the mechanism could be tested.
- Devise experiments which differentially evaluate at least two hypotheses for a given set of background conditions.
- Perform these experiments, rejecting reaction mechanisms hypotheses that are shown to be inconsistent with the experimental results.
In following this procedure, chemists do more than simply falsify: they add confirmatory power to the mechanisms that haven’t been eliminated. Indeed, in discussing similar issues, biochemist John Platt (1964) argued that good scientific inference is strong inference, whereby the goal in an experiment is to eliminate one or more hypotheses. Several contemporary philosophers have endorsed the role of eliminative induction in science (e.g., Bird 2010, Dorling 1995, Kitcher 1993, Norton 1995). It is easy to see how it can be modeled in Bayesian and other quantitative frameworks for confirmation. Specifically, as particular candidate reaction mechanisms are eliminated, the probability that one of the remaining mechanisms is correct goes up (see Earman 1992 for details).
One difficulty with eliminative induction is the source of the relevant alternative hypotheses, in this case reaction mechanisms. There is no algorithmic procedure for generating these mechanisms, and there is always the possibility that the correct mechanism has not been considered at all. This is a genuine problem, and we believe that it is the very issue that motivates chemists to turn towards falsification when thinking about mechanisms; all they can do is evaluate the plausible mechanisms that they have thought of. However, we see eliminative induction as a more plausible reflection of the epistemic situation of mechanistic chemists. This problem is not uncertainty about mechanisms compatible with experiments—chemists have evidence that weighs in favor of those. Rather, the problem is with unconceived alternatives. Structure offers one way to delineate such mechanistic possibilities: Hoffmann (1997, Chapter 29) provides a beautiful example of explicitly eliminative reasoning in his discussion of how H. Okabe and J. R. McNesby used isotopic labelling to eliminate two out of three possible mechanisms for the photolysis of ethane to ethylene. But this is an issue in all parts of science, not just mechanistic chemistry, and eliminative induction has always played a role in chemists’ reasoning about structure. How did van ’t Hoff argue for the tetrahedral carbon atom? He argued first that it was possible to account for the observed number and variety of the isomers of certain organic substances only by taking into account the arrangement of atoms in space. He then defended a tetrahedral geometry for the carbon atom by rejecting a square planar arrangement: if carbon’s geometry were square planar, there would be more isomers of substituted methane than are observed. Thus, for instance, disubstituted methane (of the form CH2X2) should have two separable isomers if it is square planar, whereas only one can be found. Assuming a tetrahedral arrangement, in contrast, would be in accord with the observed number of isomers (Brock 1992).
5.3 Logics of Discovery in Chemistry
In his classic discussion, Hans Reichenbach distinguished between the context of discovery and the context of justification. His distinction was intended to highlight the fact that we could have a logical analysis of scientific justification in the form of confirmation theory, but there could never be a logical procedure for generating hypotheses. Hypothesis generation is the creative part of science, while confirmation is the logical part. This distinction has been challenged in recent years by those that see the path of discovery contributing to justification. But chemistry provides a more interesting challenge to Reichenbach: It apparently gives us logics of discovery.
There are two subfields in which chemists sometimes speak of logics or procedures for discovery. The first is synthetic chemistry. E.J. Corey (Corey & Cheng 1989) has proposed that the synthesis of organic molecules can be rationally planned according to the logic of retrosynthetic analysis. Systematizing a long tradition in synthetic organic chemistry, Corey shows how one can reason backwards from a target molecule by finding a series of “disconnections,” bonds which one knows how to make. The resulting tree of disconnections gives potential pathways for synthesis that can then be evaluated by plausibility, or simply tried out in the laboratory.
Another area where chemists have developed a logic for discovery is in the area of drug design. Murray Goodman (Goodman & Ro 1995), for example, proposed a four-step procedure for developing candidate molecules for new medication. Say that you were interested in making a drug that would more effectively target one of the morphine receptors in the brain. You start by making a molecular analogue of morphine, perhaps with a more constrained structure. After successful synthesis, you study the molecule’s three-dimensional structure by spectroscopy and computer simulation. You then test your molecule in a biological assay to see if you have successfully targeted the receptor and to what extent. Then based on the information you get, you modify the structure, hopefully improving in each iteration.
These examples from chemistry put pressure on Reichenbach’s claim that there cannot be a logic of discovery. Moreover, they illustrate how, when a science is concerned with creating new things, procedures for discovery may become essential.
6. Chemical Reduction
One of the perennial topics in philosophy of science concerns inter-theoretic relations. In the course of debating whether biology is reducible to the physical sciences or whether psychology is reducible to biology, many philosophers assume that chemistry has already been reduced to physics. In the past, this assumption was so pervasive that it was common to read about “physico/chemical” laws and explanations, as if the reduction of chemistry to physics was complete. Although most philosophers of chemistry would accept that there is no conflict between the sciences of chemistry and physics (Needham 2010b), many would reject a stronger unity thesis. Most believe that chemistry has not been reduced to physics nor is it likely to be (see Le Poidevin 2005, for the opposite view, and Hendry & Needham 2007, for a rejoinder).
When thinking about the question of reducibility in chemistry, it is useful to break this question into two parts: The first, and more familiar one to philosophers, concerns the relationship between elements, atoms, molecules, and the fundamental particles of physics. We might ask, “Are atomic and molecular species reducible to systems of fundamental particles interacting according to quantum mechanics?” A second, less familiar question concerns the relationship between the macroscopic and microscopic descriptions of chemical substances. “Are chemical substances reducible to molecular species?” Here, the main question is whether all chemical properties that have been defined macroscopically can be redefined in terms of the properties of atoms, molecules, and their interactions.
6.1 Reduction of Molecular Species to Quantum Mechanics
Bogaard (1978), Scerri (1991, 1994) and Hendry (1998) have all questioned the possibility of fully reducing chemical theories about atoms and molecules to quantum mechanics. Bogaard argues that many key chemical concepts such as valence and bonding do not find a natural home in quantum mechanics. In a similar spirit, Scerri points out that the quantum mechanical calculations of atomic spectra standardly presented in chemistry textbooks make highly idealized assumptions about the structure of many-electron systems. These approximations are well-motivated on pragmatic grounds. However, they do not allow quantum mechanics to “approximately reduce” chemical facts, because the errors introduced by these approximations cannot be estimated (Scerri 1991, 1994). Further, one of the most important chemical trends, the length of periods in the Periodic Table, cannot be derived from quantum mechanics, unless experimentally derived chemical information is specifically introduced (Scerri 1997). Drawing on the work of Woolley (1978) and Primas (1981), Hendry (1998) argues that there are principled difficulties in accommodating molecular shape within quantum mechanics: the Born-Oppenheimer approximation effectively adds structure by hand. Although quantum chemistry can be extremely illuminating, these authors argue that it has not reduced chemistry to physics.
If one thinks that reduction means deriving the phenomenon of the higher level exclusively from the lower level, then these arguments should settle the question of reduction. More than 80 years after the discovery of quantum mechanics, chemistry has not been reduced to it. But there are two possible reductionist responses to this argument.
First, reductionists can argue that there are no principled reasons that chemical phenomena have not been derived from quantum mechanics. The problem is a lack of computational power and appropriate approximation schemes, not anything fundamental. Schwarz (2007) has made this argument against Scerri, claiming that the electronic structure of atoms, and hence the Periodic Table, is in principle derivable from quantum mechanics. He believes that quantum chemistry’s inability to reduce chemical properties is simply a manifestation of the problems shared by all of the computationally complex sciences. Debate then turns to the plausibility of such “reducibility in principle” claims.
There are also arguments that focus, at least implicitly, on chemistry’s ontology. A well-known strand of contemporary metaphysics defends physicalism, the doctrine that everything in the universe is physical (see the entry on physicalism). According to the physicalist, chemistry is “nothing but” physics, even though chemical explanations and theories are not derivable from physics. The physical world is simply composed of the fundamental particles of physics. Chemical entities and their properties have no independent reality.
The status of arguments for physicalism and the supervenience of everything on the physical are contentious within metaphysics proper, but beyond the scope of this entry. Yet we think that the failure of chemical theory to be fully derivable from physics raises interesting questions about the doctrine of physicalism. Minimally, it points to longstanding worries that the domain of the physical is not well-defined. If chemical entities such as molecules and ions end up being part of the physical ontology, one might argue that this was not a case of the reduction of chemistry to physics at all but simply the expansion of the ontology of physics to encompass the ontology of chemistry.
Independent studies of the ontology of chemistry on the basis of mereology have been undertaken by several authors (Earley 2005, Harré and Llored 2011, Needham 2010a). In disputing Scerri’s (2000, 2001, 2017b) argument against claims (Zuo et al. 1999) that orbitals have been observed, Mulder (2010) appeals to a general ontological distinction between entities, which can appropriately be said to exist, and states which don’t exist independently but are features of entities that exist. Ostrovsky (2005) and Schwarz (2006) take issue with the role of approximations in Scerri’s argument.
More controversially, some philosophers of chemistry have argued that chemical properties may constrain the behavior of physical systems, something akin to what philosophers of mind call strong emergence, or downwards causation (Kim 1999). While acknowledging the central role of quantum mechanics in understanding structure, Hendry argues that in some cases, molecular structure is an unexplained explainer. The issue arises when we consider the quantum-mechanical description of structural isomers, molecules with the same atoms, but with different molecular structures. For example, dimethyl ether and ethanol share a Hamiltonian, the quantum mechanical description of their physical states. Nevertheless, they are very different molecules. Ethanol is extremely soluble in water, whereas dimethyl ether is only partially soluble in water. Ethanol boils at 78.4°C, while dimethyl ether boils at 34.6°C. Drinking ethanol leads to intoxication, while dimethyl ether is toxic in quite different ways. Quantum mechanics can show how each of these structures is energetically stable, and illuminate how they interact with other molecules and radiation to explain the chemical and spectroscopic behaviour of ethanol and dimethyl ether, but the different structures are introduced as unexplained initial conditions. While he acknowledges that these facts are not incompatible with the claim that structure is reducible, Hendry argues that strong emergence is just as plausible an interpretation as reduction of the explanatory relationship between chemistry and quantum mechanics (2006b, 2010a).
6.2 Reduction of Substances to Molecular Species
So far we have considered intertheoretic relationships between chemistry and physics. What about within chemistry itself? Do the macroscopic and microscopic theories of chemistry align perfectly? Are all macroscopic properties of substances ultimately reducible to microscopic properties? In other words, if we have a macroscopic description of matter and a thermodynamic theory about how it behaves, can all of this be reduced to a molecular description? The answer has seemed to be “yes” to many philosophers and chemists, but philosophers of chemistry have urged caution here.
Consider first the relatively simple case of gas temperature, which has often been supposed reducible to the average kinetic energy of the gas’s molecules (cf. Nagel 1961, p. 343). A particular average kinetic energy of the molecules is only a necessary condition for having a given temperature, however. Only gases at equilibrium have a definite temperature, when all the spatial parts have the same temperature as the whole (reflecting the fact that temperature is an intensive property). A sufficient condition would need to complement the average kinetic energy with a microscopic correlate of the macroscopic condition of being at thermodynamic equilibrium. Statistical mechanics specifies the relevant correlative condition as that of the energy being distributed over the gas molecules in accordance with the Boltzmann distribution. But the Boltzmann distribution is expressed as a function of the temperature, and its derivation from Boltzmann’s microscopic construal of entropy appeals to the thermodynamic law connecting entropy with temperature. Accordingly, the necessary and sufficient microscopic condition for gas temperature becomes circular when construed as a reduction of the concept of temperature (Needham 2010b; Bishop 2010)
Although the reduction of temperature to microscopic properties is problematic, it is a relatively easy candidate for reduction. Properties concerned with chemical changes such as phase transitions, solubility, and reactivity, are considerably more complex. As we discussed in Section 4.5, a purely microscopic description of matter is not coextensive with all chemical properties. Solubility, for example, is not fully explained by microscopic properties. While we can explain in rough qualitative fashion that substances dissolve when their ions or molecules have more affinity for the solvent than they do for each other, this doesn’t recover the subtle, quantitative features of solubility. It also leaves the solubility of nonionic substances untouched. Predicting these features requires appeals to thermodynamics, and the alleged reduction of thermodynamics to statistical mechanics is considered highly contentious (Sklar 1993).
As we have seen in this case, even very fruitful applications of physical and chemical theory at the microscopic level are often insufficient to reduce chemically important properties. Whether the general notion of chemical substance, or the property of being a particular substance for each of the millions of known substances, can be reduced to microstructure needs to be demonstrated and not merely assumed. While there is no in-principle argument that reductions will always be impossible, essential reference is made back to some macroscopically observable chemical property in every formal attempt of reduction that we are aware of. In the absence of definite arguments to the contrary, it seems reasonable to suppose that chemistry employs both macroscopic and microscopic concepts in detailed theories which it strives to integrate into a unified view. Although plenty of chemistry is conducted at the microscopic level alone, macroscopic chemical properties continue to play important experimental and theoretical roles throughout chemistry.
In the background of all of these debates about chemical reduction are issues concerning the criteria for successful reduction. All of the literature that we have discussed make explicit or implicit reference to Nagel’s influential account of reduction. Beyond the philosophy of chemistry literature, this account has also been presupposed by critics of particular reductionist theses (e.g. Davidson 1970), even when making points about the inapplicability of Nagel’s account to particular sciences (Kitcher 1984). But Nagel’s account of reduction is thought by many to be unrealistic and inapplicable to actual science because of the logical requirements it assumes.
Perhaps part of the anti-reductionist consensus in the philosophy of chemistry literature is driven by the stringent demands of Nagel’s account. But even if Nagel’s account is weakened to allow approximative arguments (as Hempel modified his DN model of explanation), as some advocates of reductionism have urged (e.g., Schaffner 1967; Churchland 1985), this still doesn’t circumvent the problem of the appeal to macroscopic properties in the explanation of microscopic properties. Current chemical theory makes essential reference to both microscopic and macroscopic chemical concepts with both chemical and quantum mechanical origins. We know of no convincing substantial examples where either of these aspects have been entirely excised.
7. Modeling and Chemical Explanation
Almost all contemporary chemical theorizing involves modeling, the indirect description and analysis of real chemical phenomena by way of models. From the 19th century onwards, chemistry was commonly taught and studied with physical models of molecular structure. Beginning in the 20th century, mathematical models based on classical and quantum mechanics were successfully applied to chemical systems. This section discusses some of the philosophical questions that arise when we consider modeling in chemistry more directly.
7.1 Physical Modeling
Chemistry’s modeling tradition began with physical models of atoms and molecules. In contemporary chemical education, much emphasis is placed on the construction and manipulation of such models. Students in organic chemistry classes are often required to purchase plastic molecular modeling kits, and it is not uncommon to see complex molecular structures built from such kits in professional laboratory settings.
The use of molecular models gained special prominence in the middle of the 19th, helping chemists to understand the significance of molecular shape (Brock 2000). While such structures could be represented on paper, physical models gave an immediacy and an ease of visualization that sketches alone did not provide. In the middle of the twentieth century, the discovery of the double helical structure of DNA was aided by the manipulation of physical models (Watson 1968).
7.2. Mathematical Modeling
While physical modeling has been important historically, and is still a central part of chemical education and some investigations in stereochemistry, contemporary chemical models are almost always mathematical. Families of partially overlapping, partially incompatible models such as the valence bond, molecular orbital, and semi-empirical models are used to explain and predict molecular structure and reactivity. Molecular mechanical models are used to explain some aspects of reaction kinetics and transport processes. And lattice models are used to explain thermodynamic properties such as phase. These and other mathematical models are ubiquitous in chemistry textbooks and articles, and chemists see them as central to chemical theory.
Chemists are very permissive about which kinds of mathematical structures can serve as models. But while just about any kind of mathematical structure can serve as a chemical model, different types of systems lend themselves to particular kinds of mathematical structures used in modeling. For example, the most common kinds of mathematical structures employed in quantum chemistry are state spaces, which typically correlate sub-molecular particle distances with the total energy of chemical systems. Other parts of chemical modeling are dynamic, hence they employ trajectory spaces, which can represent the course of a reaction over time. Still other kinds of mathematical structures such as graphs and groups can be employed to model molecular structure and symmetry.
The purpose of many exercises in chemical modeling is to learn about real systems. In these cases, the model must bear certain relationships to real-world systems. But these relationships needn’t always be of extremely high fidelity. For example, Linus Pauling (1939) and early proponents of the simple valence bond model believed that this model captured the essential physical interactions that give rise to chemical bonding. This method is closely related to Lewis’ conception of bonding, treating molecules as composed of atomic cores (nuclei together with inner-shell electrons) and valence electrons which give rise to localized bonds. It stands in contrast to the molecular orbital method, which doesn’t localize the bonding electrons to any particular part of the molecule. Modern quantum chemists think of the valence bond model as a template for building models of greater complexity. Thus if a modern quantum chemist deploys the simple valence bond model to study a real molecule, she does so with a much lower standard of fidelity than Pauling would have. Her use of the model is only intended to give a first approximation to the most important features of the system.
7.3. Modeling and Explanation
Much of contemporary theoretical research in chemistry involves the application of quantum mechanics to chemistry. While exact solutions to the quantum mechanical descriptions of chemical phenomena have not been achieved, advances in theoretical physics, applied mathematics, and computation have made it possible to calculate the chemical properties of many molecules very accurately and with few idealizations. The approach of striving for ever more accurate calculations with decreasing levels of idealization is endorsed by many quantum chemists. For example, the development team of Gaussian, one of the leading packages for doing quantum chemical calculations, explicitly endorses this position. While they admit that there are many considerations that enter into the choice of the degree of approximation or “level of theory” for any calculation, the goal is to de-idealize the models as much as possible. They argue that quantum chemical calculations which are arbitrarily close to the exact solutions are the “limit to which all approximate methods strive” (Foresman & Frisch 1996).
This method of developing chemical theory relies on a systematic refinement of theories, attempting to bring them closer to the truth. Philosophers of science have called this process Galilean idealization, because as in Galileo’s work, idealizations are introduced for reasons of tractability and are removed as soon as possible (McMullin 1985; Weisberg 2007b). But not all chemists have shared this focus on ever more accurate calculations. Reflecting on why he didn’t choose this path in his own career, theorist Roald Hoffmann wrote:
I took a different turn, moving from being a calculator … to being an explainer, the builder of simple molecular orbital models … [and] I feel that actually there is a deeper need than before for [this] kind of work … analyzing a phenomenon within its own discipline and seeing its relationship to other concepts of equal complexity (1998).
Elsewhere in this article, Hoffmann admits that quantum chemistry is enormously successful in its predictive power, and continues to give us better approximations to the fundamental theory. Yet the attitude expressed in this paragraph seems to be that simple, idealized models are needed for chemical theorizing. Thus, the central philosophical question arises: Given the availability of models that are closer to the truth, why work with idealized ones?
One answer is given by Felix Carroll, a physical organic chemist:
Why then do not we just talk about high-level theoretical calculations and ignore the simple theory? We must choose the model that is sufficiently accurate for our computational purposes, yet still simple enough that we have some understanding of what the model describes. Otherwise, the model is a black box, and we have no understanding of what it does, perhaps even no idea whether the answers it produces are physically reasonable (Carroll 1998).
Carroll does not elaborate on these issues, but this passage contains the central message: Simple models prevent our theories from having a “black-box” character, meaning that they will not simply be a recipe for calculating without giving any physical insight. Carroll claims that simple models are necessary in order to expose the mechanisms by which chemical phenomena come about. High-level theoretical calculations are not capable of showing us these mechanistic relationships, even though they are based on the quantum mechanical principles that describe the fundamental physics of the system. Or, as Hoffmann puts the point: “[I]f understanding is sought, simpler models, not necessarily the best in predicting all observables in detail, will have value. Such models may highlight the important causes and channels” (Hoffmann, Minkin, & Carpenter 1996).
Why should it be the case that simple models have less black-box character than others? One explanation appeals to our cognitive limitations. We can only hold a couple of steps of an argument in our mind at once. Modern, high-level calculations can take hours or days to compute using fast computers. Even if every step was made explicit by the computer, it would be impossible to hold the calculational steps in mind and hence hard to understand the reason for the result, even if one was convinced that the answer was correct. Paul Humphreys has called this the epistemic opacity of simulations (2004).
There is a second reason for employing simple, more highly idealized models in chemistry, which stems from the explanatory traditions of chemistry. In developing this point, Hoffmann argues that there are two modes of explanation that can be directed at chemical systems: horizontal and vertical (Hoffmann 1997). Vertical explanations are what philosophers of science call deductive nomological explanations. These explain a chemical phenomenon by deriving its occurrence from quantum mechanics. Calculations in quantum chemistry are often used to make predictions, but insofar as they are taken to explain chemical phenomena, they follow this pattern. By showing that a molecular structure is stable, the quantum chemist is reasoning that this structure was to be expected given the underlying physics.
In contrast with vertical mode, the horizontal mode of explanation attempts to explain chemical phenomena with chemical concepts. For example, all first year organic chemistry students learn about the relative reaction rates of different substrates undergoing the SN2 reaction. An organic chemist might ask “Why does methyl bromide undergo the SN2 reaction faster than methyl chloride?” One answer is that “the leaving group Br− is a weaker base than Cl−, and all things being equal, weaker bases are better leaving groups.” This explains a chemical reaction by appealing to a chemical property, in this case, the weakness of bases.
Hoffmann doesn’t say much about the differing value of the horizontal and vertical explanations, but one important difference is that they give us different kinds of explanatory information. Vertical explanations demonstrate that chemical phenomena can be derived from quantum mechanics. They show that, given the (approximate) truth of quantum mechanics, the phenomenon observed had to have happened. Horizontal explanations are especially good for making contrastive explanations, which allows the explanation of trends. Consider again our example of the rate of an SN2 reaction. By appealing to the weakness of Br− as a base, the chemist invokes a chemical property, shared across other molecules. This allows her to explain methyl bromide’s reactivity as compared to methyl chloride, and also methyl fluoride, methyl iodide, etc. Insofar as chemists want to explain trends, they make contrastive explanations using chemical concepts.
Reflecting on the nature of chemical theorizing, the eminent chemical theorist Charles Coulson (1910–1974) makes a similar point. He wrote:
[T]he role of quantum chemistry is to understand these concepts and show what are the essential features in chemical behavior. [Chemists] are anxious to be told … why the H–F bond is so strong, when the F–F bond is so weak. They are content to let spectroscopists or physical chemists make the measurements; they expect from the quantum mechanician that he will explain why the difference exists. … So the explanation must not be that the computer shows that [the bonds are of different length], since this is not an explanation at all, but merely a confirmation of experiment (Coulson 1960, p. 173).
Although Coulson, Carroll, and Hoffmann defend the use of simple, idealized models to generate horizontal explanations, it is not clear that quantum calculations can never generate contrastive explanations. Although single vertical explanations are not contrastive, a theorist can conduct multiple calculations and in so doing, generate the information needed to make contrastive explanations. Many of the best examples of quantum chemistry have this character: a series of closely related calculations, attempting to get at chemically relevant trends.
Bibliography
- Aristotle [DG], On Generation and Corruption (De Generatione et Corruptione), in The Complete Works of Aristotle (Volume 1), J. Barnes (ed.), Princeton: Princeton University Press, 1984, 512–554.
- Aristotle, On the Heavens (De Caelo), in The Complete Works of Aristotle (Volume 1), J. Barnes (ed.), Princeton: Princeton University Press, 1984, 447–511.
- Atkins, P.W., 1991, The Second Law, New York: Freeman.
- Bader, R.F.W., 1991, Atoms in Molecules, Oxford: Clarendon Press.
- –––, 2009, “Bond Paths are Not Chemical Bonds,” Journal of Physical Chemistry A, 113: 10391–10396.
- Banchetti, Marina P., 2020, Chemical Philosophy of Robert Boyle: Mechanicism, Chymical Atoms, and Emergence, Oxford: Oxford University Press.
- Berson, J., 2008, “Molecules with Very Weak Bonds: The Edge of Covalency” Philosophy of Science, 75: 947–957.
- Berzelius, Jacob, 1813, “Experiments on the Nature of Azote, of Hydrogen, and of Ammonia, and Upon the Degrees of Oxidation of Which Azote is Susceptible”, Annals of Philosophy, 2: 276–86, 357–68.
- –––, 1815, “An Address to those Chemists Who Wish to Examine the Laws of Chemical Proportions,” Annals of Philosophy, 5: 122–131.
- Biggs, Norman L., E. Keith Lloyd and Robin J. Wilson, 1976, Graph Theory 1736–1936, Oxford: Clarendon Press.
- Bird, Alexander, 2010, “Eliminative Induction: Examples from medicine”, Studies in History and Philosophy of Science, 41: 345–52.
- Bishop, Robert C., 2010, “Whence Chemistry?”, Studies in History and Philosophy of Modern Physics, 41: 171–177.
- Bogaard, Paul A., 1978, “The Limitations of Physics as a Chemical Reducing Agent”, Proceedings of the Philosophy of Science Association (PSA 1978), 2: 345–56.
- Bréhier, Émile, 1951, Chrysippe et l’ancien stoïcisme, 2nd edition, Presses Universitaires de France, Paris.
- Brock, W.H., 2000, The Chemical Tree: A History of Chemistry, New York: W.H. Norton.
- Brush, Stephen, 1999a, “Dynamics of Theory Change in Chemistry: The Benzene Problem 1865–1945”, Studies in History and Philosophy of Science, 30: 21–79.
- –––, 1999b, “Dynamics of Theory Change in Chemistry: Benzene and Molecular Orbitals, 1945–1980”, Studies in History and Philosophy of Science, 30: 263–302.
- Burrau, Øjvind, 1927, “Berechnung des Energiewertes des Wasserstoffmolekel-Ions (H2+)”, Videnskabernes Selskab Matematisk-Fysiske Meddelelser, 7: 1–18.
- Bursten, Julia, 2014, “Microstructure without Essentialism: A New Perspective on Chemical Classification”, Philosophy of Science, 81: 633–53.
- Carpenter, B., 1984, Determination of Organic Reaction Mechanisms, New York: John Wiley and Sons.
- Carroll, F. A., 1998, Perspectives on Structure and Mechanism in Organic Chemistry, Pacific Grove: Brooks/Cole.
- Cerpa, Erick, A. Krapp, A. Vela, and G. Merino, 2008, “The Implications of the External Symmetry on the External Potential on Bond Paths,” Chemistry: A European Journal, 14: 10232–10234.
- Chalmers, Alan, 1993, “The Lack of Excellency of Boyle’s Mechanical Philosophy”, Studies in History and Philosophy of Science, 24: 541–564.
- –––, 2002, “Experiment Versus Mechanical Philosophy in the Work of Robert Boyle: A Reply to Anstey and Pyle”, Studies in History and Philosophy of Science, 33: 191–7.
- –––, 2005a, “Atomism from the 17th to the 20th Century”, The Stanford Encyclopedia of Philosophy (Fall 2005 Edition), Edward N. Zalta(ed.), URL = <https://plato.stanford.edu/archives/fall2005/entries/atomism-modern/>.
- –––, 2005b, “Transforming Atomic Chemistry into an Experimental Science: The Limitations of Dalton’s Theory”, The Rutherford Journal, 1 [Available online].
- –––, 2008, “Atomism and Aether in Nineteenth-Century Physical Science”, Foundations of Chemistry, 10: 157–66.
- –––, 2009, The Scientist’s Atom and the Philosopher’s Stone: How Science Succeeded and Philosophy Failed to Gain Knowledge of Atoms (Boston Studies in the Philosophy of Science, Vol. 279), Dordrecht: Springer.
- Chang, Hasok, 2012, Is Water H2O? Evidence, Realism and Pluralism (Boston Studies in the Philosophy of Science, Vol. 293), Dordrecht: Springer.
- Churchland, P, 1985, “Reduction, Qualia, and Direct Introspection,” Journal of Philosophy, 82: 8–28.
- Cooper, J., 2004, “Two notes on Aristotle on mixture”, In J. Cooper, Knowledge, nature, and the good, Princeton: Princeton University Press.
- Corey, E. J. and X. Cheng, 1989, The Logic of Chemical Synthesis, New York: Wiley.
- Coulson, C. A., 1952, “What Is a Chemical Bond?”, Scientific Journal of the Royal College of Science, 21: 11–29.
- –––, 1960, “Present state of molecular structure calculations”. Reviews of Modern Physics, 32: 170–177.
- Crum Brown, Alexander, 1864 [1865], “On the Theory of Isomeric Compound,” Transactions of the Royal Society of Edinburgh 23, 707–19. Page references are to the 1865 reprint in the Journal of the Chemical Society, 18: 230–45.
- Davidson, Donald, 1970, “Mental Events”, in L. Foster and J. W. Swanson (eds.), Experience and Theory, Duckworth, London; reprinted in Davidson 1980, pp. 207–25.
- –––, 1980, Essays on Actions and Events, New York: Oxford University Press.
- Deltete, Robert J., 2000, The Historical Development of Energetics, trans. of G. Helm, Die Energetik nach ihrer geschichtlichen Entwicklung, Veit & Comp., Leipzig, 1898, by Kluwer, Dordrecht.
- Dorling, J., 1995, “Einstein’s Methodology of Discovery was Newtonian Deduction from the Phenomena”, in J. Leplin (ed.), The Creation of Ideas in Physics, Dordrecht: Kluwer, 97–112.
- Duhem, Pierre, 1892, “Notation atomique et hypothèses atomistiques”, Revue des questions scientifiques, 31, 391–457; translated (with the original pagination) by Paul Needham as “Atomic Notation and Atomistic Hypotheses”, Foundations of Chemistry, 2 (2000): 127–180.
- –––, 1902, Le mixte et la combinaison chimique: Essai sur l’évolution d’une idée, C. Naud, Paris; reprinted Fayard, Paris, 1985. Translated in Duhem (2002).
- –––, 2002, Mixture and Chemical Combination, and Related Essays, translated and edited by Paul Needham, Kluwer, Dordrecht.
- Earley, Joseph E., 2005, “Why There is no Salt in the Sea”, Foundations of Chemistry, 7: 85–102.
- Earman, J., 1992, Bayes or Bust? Cambridge, MA: MIT Press.
- Einstein, Albert, 1905 [1956], Investigations on the Theory of the Brownian Movement, Dover Books, New York.
- Emerton, Norma E., 1984, The Scientific Reinterpretation of Form, Cornell University Press, Ithaca, New York.
- Feynman, R.P., 1939, “Forces in Molecules”, Physical Review, 56: 340–3.
- Fine, K., 1995, “The problem of mixture”, Pacific Philosophical Quarterly, 76: 266–369.
- Finney, John L., 2004, “Water? What’s so special about it?”, Philosophical Transactions of the Royal Society of London B, 359: 1145–1165.
- Foresman, J. B., & Frisch, A., 1996, Exploring chemistry with electronic structure methods, second edition, Pittsburgh: Gaussian, Inc.
- Fox, Robert, 1971, The Caloric Theory of Gases: From Lavoisier to Regnault, Clarendon Press, Oxford.
- Geoffroy, Etienne François, 1718 [1996], “Table of Different Relations Observed in Chemistry Between Different Substances”, Science in Context, 9: 313–20.
- Gibbs, J. W., 1948, “On the equilibrium of heterogeneous substances”, in The collected works of J. Willard Gibbs, Vol. I (pp. 55–353), Yale University Press, New Haven. (First published in Transactions of the Connecticut Academy of Arts and Sciences, 3: 1876–1878, 108–248, 343–520; this edition was first published in 1906).
- Gillespie, Ronald J. and Paul L. A. Popelier, 2001, Chemical Bonding and Molecular Geometry: From Lewis to Electron Densities, Oxford University Press, Oxford.
- Goodman, M and S. Ro, 1995, “Peptidomimetics for Drug Design,” Burger’s Medicinal Chemistry and Drug Discovery, Vol. I (Eds.: M. E. Wolff), John Wiley & Sons, Inc., 803–861.
- Goodwin, W., 2012, “Mechanisms and Chemical Reactions,” Handbook of the Philosophy of Science, Vol 6: Philosophy of Chemistry, R. Hendry, P. Needham, and A. Woody (eds.), Amsterdam: Elsevier, 301–327.
- Harré, Rom and Jean-Pierre Llored, 2011, “Mereologies as the Grammars of Chemical Discourses”, Foundations of Chemistry, 13: 63–76.
- Havstad, Joyce C., 2018, “Messy Chemical Kinds”, British Journal for the Philosophy of Science, 69: 719–743
- Hempel, C. G., 1965, “Aspects of Scientific Explanation”, Aspects of Scientific Explanation, Free Press, Collier-Macmillan, Toronto; pp. 331–496.
- Hendry, Robin Findlay, 1998, “Models and Approximations in Quantum Chemistry”, in Niall Shanks (ed.), Idealization IX: Idealization in Contemporary Physics (Poznan Studies in the Philosophy of the Sciences and the Humanities, Vol. 63), Amsterdam: Rodopi; 123–42.
- –––, 2005, “Lavoisier and Mendeleev on the Elements”, Foundations of Chemistry, 7: 31–48.
- –––, 2006a, “Elements, compounds and other chemical kinds”, Philosophy of Science, 73: 864–75.
- –––, 2006b, “Is There Downwards Causation in Chemistry?”, in Davis Baird, Eric Scerri and Lee McIntyre, eds., Philosophy of Chemistry: Synthesis of a New Discipline (Boston Studies in the Philosophy of Science), Dordrecht: Springer, 173–89.
- –––, 2008, “Two Conceptions of the Chemical Bond,” Philosophy of Science, 75(5): 909–920.
- –––, 2010a, “Ontological Reduction and Molecular Structure”, Studies in History and Philosophy of Modern Physics, 41: 183–191.
- –––, 2010b, “The chemical bond: structure, energy and explanation,” in Mauro Dorato, Miklos Redei and Mauricio Suarez (eds.), EPSA: Philosophical Issues in the Sciences: Launch of the European Philosophy of Science Association, Berlin: Springer, 117–27.
- –––, 2010c, “Entropy and chemical substance”, Philosophy of Science, 77: 921–32.
- –––, 2016, “Natural kinds in chemistry,” in Grant Fisher and Eric Scerri (eds.), Essays in the Philosophy of Chemistry, Oxford: Oxford University Press, 253–75.
- Hendry, Robin Findlay and Paul Needham, 2007, “Le Poidevin on the Reduction of Chemistry”, British Journal for the Philosophy of Science, 58: 339–53.
- Hoffmann, R., 1997, The Same and Not the Same, New York: Columbia University Press.
- –––, 1998, “Qualitative thinking in the age of modern computational chemistry–or what Lionel Salem knows”. Journal of Molecular Structure, 424: 1–6.
- Hoffmann, R., Minkin, V. I., & Carpenter, B. K., 1996, “Ockham’s razor and chemistry”. Bulletin de la Société Chimique de France, 133: 117–130.
- Humphreys, P., 2004, Extending Ourselves, New York: Oxford University Press.
- Kitcher, Philip, 1984, “1953 and All That. A Tale of Two Sciences”, Philosophical Review, 93: 335–73.
- –––, 1993, The Advancement of Science, New York: Oxford University Press.
- Kim, Jaegwon, 1999, “Making Sense of Emergence”, Philosophical Studies, 95: 3–36.
- Kekulé A., 1858 [1963], Uber die Constitution und die Metamorphosen der chemischen Verbindungen und uber die chemische Natur des Kohlenstoffs. Ann. Chem. Pharm., 106: 129–159. Page references are to the translation as “The constitution and metamorphoses of chemical compounds and the chemical nature of carbon,” in Theodor Benfey (ed.) Classics in the theory of chemical combination, New York: Dover, 109–131.
- Klein, Ursula, 1994, “Origin of the Concept of Chemical Compound”, Science in Context, 7: 163–204.
- –––, 1995, “E. F. Geoffroy’s Table of Different ‘Rapports’ Between Different Chemical Substances—A Reinterpretation”, Ambix, 42: 79–100.
- –––, 2003, Experiments, models and paper tools: Cultures of organic chemistry in the nineteenth century, Stanford: Stanford University Press.
- Kohler, Robert, 1971, “The Origin of G.N. Lewis’s Theory of the Shared Pair Bond,” Historical Studies in the Physical Sciences, 3: 343–376.
- –––, 1975, “G.N. Lewis’s Views on Bond Theory, 1900–16.” British Journal for the History of Science, 8: 233–239.
- Kossel, Walther, 1916, “Uber Molkulbildung als Frage der Atombau”, Annalen der Physik, 49: 229–362.
- Kragh, Helge, 2000, “Conceptual Changes in Chemistry: The Notion of a Chemical Element, ca. 1900–1925” Studies in History and Philosophy of Modern Physics 31B: 435–50.
- –––, 2002, “The Vortex Atom: A Victorian Theory of Everything”, Centaurus, 44: 32–126.
- Kripke, Saul, 1980, Naming and Necessity, Oxford: Blackwell.
- Ladyman, James, 2011, “Structural Realism versus Standard Scientific Realism: The Case of Phlogiston and Dephlogisticated Air”, Synthese, 180: 87–101.
- Lavoisier, Antoine–Laurent, 1789 [1965], Elements of Chemistry in a new systematic order, containing all the modern discoveries, trans. by Robert Kerr of Traité Élémentaire de Chimie, reprinted New York: Dover.
- Le Poidevin. R., 2005, “Missing Elements and Missing Premises: A Combinatorial Argument for the Ontological Reduction of Chemistry”, British Journal for the Philosophy of Science, 56: 117–134
- Lewis, G. N., 1916, “The Atom and the Molecule”, Journal of the American Chemical Society, 38: 762–85.
- –––, 1923 [1966], Valence and the structure of atoms and molecules, New York: Dover.
- Machamer, P., L. Darden, and C. Craver, 2000, “Thinking About Mechanisms”, Philosophy of Science, 67: 1–25.
- Maiocchi, Roberto, 1990, “The Case of Brownian Motion”, British Journal for the History of Science, 23: 257–83.
- Malt, Barbara, 1994, “Water is Not H2O”, Cognitive Psychology, 27: 41–70
- Mátyus, Edit, Jürg Hutter, Ulrich Müller-Herold and Markus Reiher, 2011, “On the Emergence of Molecular Structure”, Physical Review A, 83: 052512.
- McMullin, E., 1985, “Galilean Idealization”, Studies in History and Philosophy of Science, 16: 247–73.
- Mendeleev, Dmitri, 1869 [2005], “On the correlation between the properties of the elements and their atomic weights,” in William B. Jensen (ed.), Mendeleev on the Periodic Law: Selected Writings, 1869–1905, New York: Dover, 18–37.
- Meinel, Christoph, 1988, “Early Seventeenth-century Atomism: Theory, Epistemology, and the Insufficiency of Experiment”, Isis, 79: 68–103.
- –––, 2004, “Molecules and Croquet Balls,” in Models: The Third Dimension of Science, Soraya de Chadarevian and Nick Hopwood (eds.), Stanford: Stanford University Press, pp. 242–275.
- Mulder, Peter, 2010, “On the Alleged Non-Existence of Orbitals”, Studies in History and Philosophy of Modern Physics, 41: 178–82.
- Mulliken, R.S., 1931, “Bonding Power of Electrons and Theory of Valence”, Chemical Reviews, 9: 347–88.
- Nagel, Ernest, 1961, The Structure of Science, London: Routledge and Kegan Paul.
- Needham, Paul, 1996, “Substitution: Duhem’s Explication of a Chemical Paradigm”, Perspectives on Science, 4: 408–33.
- –––, 2000, “What is Water?”, Analysis, 60: 13–21.
- –––, 2004, “When did atoms begin to do any explanatory work in chemistry?”, International Studies in the Philosophy of Science, 8: 199–219.
- –––, 2008, “Is Water a Mixture?—Bridging the Distinction Between Physical and Chemical Properties”, Studies in History and Philosophy of Science, 39: 66–77.
- –––, 2009a, “An Aristotelian Theory of Chemical Substance”, Logical Analysis and History of Philosophy, 12: 149–64.
- –––, 2009b, “Reduction and Emergence: A Critique of Kim”, Philosophical Studies, 146: 93–116.
- –––, 2010a, “Substance and Time”, British Journal for the Philosophy of Science, 61: 485–512.
- –––, 2010b, “Nagel’s Analysis of Reduction: Comments in Defence as Well as Critique”, Studies in History and Philosophy of Modern Physics, 41: 163–170.
- –––, 2011, “Microessentialism: What is the Argument?”, Noûs, 45(1): 1–21.
- –––, 2014, “The Source of Chemical Bonding”, Studies in History and Philosophy of Science, 45: 1–13.
- –––, 2017a, “Determining Sameness of Substance”, British Journal for the Philosophy of Science, 68(4): 953–79.
- –––, 2017b, Macroscopic Metaphysics: Middle-Sized Objects and Longish Processes, Cham: Springer.
- Newman, William R., 1996, “The Alchemical Sources of Robert Boyle’s Corpuscular Philosophy”, Annals of Science, 53: 567–85.
- Norton, J., 1995, “Eliminative Induction as a Method of Discovery”, in J. Leplin (ed.), The Creation of Ideas in Physics, Dordrecht: Kluwer, 29–69.
- Nye, Mary Jo, 1972, Molecular Reality, New York: Elsevier, 1972.
- Ostrovsky, V. N., 2005, “Towards a Philosophy of Approximations in the ‘Exact’ Sciences”, Hyle: International Journal for the Philosophy of Chemistry, 11: 101–26.
- Ostwald, Wilhelm, 1904, “Elements and Compounds”, Journal of the Chemical Society, 85: 506–22.
- Paneth, F.A., 1962, “The Epistemological Status of the Chemical Concept of Element”, British Journal for the Philosophy of Science, 13: 1–14, 144–160.
- Pauling, L., 1939, The Nature of the Chemical Bond, Ithaca: Cornell University Press.
- Perrin, Jean, 1913 [1990], Atoms, Ox Bow Press, Woodbridge, Connecticut.
- Platt, J., 1964, “Strong Inference”, Science, 146: 347–353.
- Primas, H., 1981, Chemistry, Quantum Mechanics and Reductionism, New York: Springer-Verlag.
- Putnam, Hilary, 1975, “The Meaning of ‘Meaning’,” in Philosophical Papers, Vol. 2, Cambridge: Cambridge University Press, pp. 215–71.
- –––, 1990, “Is water necessarily H2O?” in H. Putnam, Realism with a Human Face, Cambridge, MA: Harvard University Press, 54–79.
- Ramberg, P., 2003, Chemical Structure, Spatial Arrangement: The Early History of Stereochemistry, 1874–1914, Aldershot: Ashgate.
- Ramsey, J., 1997, “Molecular Shape, Reduction, Explanation and Approximate Concepts”, Synthese 111: 233–251.
- Ritter, Christopher, 2001, “An Early History of Alexander Crum Brown’s Graphical Formulas,” in Tools and Modes of Representation in the Laboratory Sciences, Ursula Klein (ed.), Dordrecht: Kluwer, pp. 35–46.
- Rocke, Alan J., 1984, Chemical Atomism in the Nineteenth Century: From Dalton to Cannizzaro, Columbus, OH: Ohio State University Press.
- –––, 2010, Image and Reality: Kekulé, Kopp and the Scientific Imagination, Chicago: University of Chicago Press.
- Russell, Colin A., 1971, The History of Valency, Leicester: Leicester University Press.
- Scerri, Eric, 1991, “The Electronic Configuration Model, Quantum Mechanics and Reduction”, British Journal for the Philosophy of Science, 42: 309–25
- –––, 1994, “Has Chemistry Been at Least Approximately Reduced to Quantum Mechanics?”, in D. Hull, M. Forbes and R. Burian (eds.), PSA 1994 (Vol. 1), Philosophy of Science Association.
- –––, 1997, “The Periodic Table and the Electron”, American Scientist, 85: 546–553.
- –––, 2000, “Have Orbitals Really Been Observed?”, Journal of Chemical Education, 77 (11): 1492–4.
- –––, 2006, The Periodic Table: Its Story and its Significance, Oxford: Oxford University Press.
- Schaffner, K., 1967, “Approaches to Reduction,” Philosophy of Science, 34: 137–147.
- Schwarz, W. H. Eugen, 2007, “Recommended Questions on the Road Towards a Scientific Explanation of the periodic System of Chemical Elements with the Help of the Concepts of Quantum Physics”, Foundations of Chemistry, 9: 139–88.
- –––, 2006, “Measuring Orbitals: Provocation or Reality?”, Angewandte Chemie International Edition, 45 (10): 1508–17.
- Seifert, Vanessa A., 2020a, “The Role of Idealisations in Describing an Isolated Molecule”, Foundations of Chemistry, 22(1): 15–29.
- –––, 2020b, “The Strong Emergence of Molecular Structure”, European Journal for Philosophy of Science, 10(3): 1–25
- –––, 2023, “The Chemical Bond is a Real Pattern” Philosophy of Science, 90(2): 269–287.
- Seifert, Vanessa A. and Alexander Franklin, 2024, “The Problem of Molecular Structure Just is the Measurement Problem”, The British Journal for the Philosophy of Science, 75(1): 31–59.
- Sklar, Lawrence, 1993, Physics and Chance: Philosophical Issues in the Foundations of Statistical Mechanics, Cambridge: Cambridge University Press.
- Sutcliffe, B.T. and R.G. Woolley, 2012, “Atoms and Molecules in Classical Chemistry and Quantum Mechanics”, Handbook of the Philosophy of Science, Vol 6: Philosophy of Chemistry, R. Hendry, P. Needham, and A. Woody (eds.), Amsterdam: Elsevier, 387–426.
- Todd, Robert B., 1976, Alexander of Aphrodisias on Stoic Physics, Leiden: E. J. Brill.
- van Brakel, Jaap, 1986, “The Chemistry of Substances and the Philosophy of Mass Terms”, Synthese, 69: 291–324.
- –––, 1997, “Chemistry as the Science of the Transformation of Substances”, Synthese, 111: 253–82.
- –––, 2000a, Philosophy of Chemistry: Between the Manifest and the Scientific Image, Leuven: Leuven University Press.
- –––, 2000b, “The Nature of Chemical Substances”, in N. Bhushan and S. Rosenfeld (eds.), Of Minds and Molecules, Oxford: Oxford University Press, pp. 162–84.
- Watson, James D., 1968, The Double Helix: A Personal Account of the Discovery of the Structure of DNA, New York: New American Library.
- Weininger, Stephen J., 2014, “Reactivity and its Contexts”, in Ursula Klein and Carsten Reinhardt (eds.), Objects of Chemical Inquiry, Leiden: Brill, 203–36.
- Weisberg, M., 2004, “Qualitative theory and chemical explanation”. Philosophy of Science, 71: 1071–1081.
- –––, 2005, “Water is Not H2O,” Philosophy of Chemistry: Synthesis of a New Discipline, D. Baird, et al. (eds.), New York: Springer, 337–345.
- –––, 2007a, “Who is a modeler?” British Journal for Philosophy of Science, 58: 207–233.
- –––, 2007b, “Three Kinds of Idealization” Journal of Philosophy, 104 (12): 639–659.
- –––, 2008, “Challenges to the Structural Conception of Bonding,” Philosophy of Science, 75: 932–946.
- Weisheipl, James A., 1963, “The Concept of Matter in Fourteenth Century Science”, in Ernan McMullin (ed.), The Concept of Matter in Greek and Medieval Philosophy, Notre Dame: University of Notre Dame Press.
- Wikforss, Åsa, 2010, “Are Natural Kind Terms Special?”, in H. Beebee and N. Sabbarton-Leary (eds.), The Semantics and Metaphysics of Natural Kinds, New York: Routledge, pp. 64–83.
- Wood, R. and M. Weisberg, 2004, “Interpreting Aristotle on mixture: problems about elemental composition from Philoponus to Cooper”, Studies in the History and Philosophy of Science, 35: 681–706.
- Woody, A.I., 2000, “Putting Quantum Mechanics to Work in Chemistry: The Power of Diagrammatic Representation”, Philosophy of Science, 67: S612–27.
- –––, 2012, “Concept Amalgamation and Representation in Quantum Chemistry”, Handbook of the Philosophy of Science (Volume 6: Philosophy of Chemistry), R. Hendry, P. Needham, and A. Woody (eds.), Amsterdam: Elsevier, 427–66.
- Woolley, R., 1978, “Must a Molecule Have a Shape?”, Journal of the American Chemical Society, 100: 1073–1078.
- Zuo, J. M., M. Kim, M. O’Keeffe and J. C. H. Spence, 1999, “Direct Observation of d-orbital Holes and Cu-Cu Bonding in Cu2O”, Nature, 401 (6748): 4952.
Academic Tools
How to cite this entry. Preview the PDF version of this entry at the Friends of the SEP Society. Look up topics and thinkers related to this entry at the Internet Philosophy Ontology Project (InPhO). Enhanced bibliography for this entry at PhilPapers, with links to its database.


Acknowledgments
The authors would like to thank Brad Berman, Mark Goodwin, Roald Hoffmann, Deena Skolnick Weisberg, and the Stanford Encyclopedia editors for extremely helpful comments and advice. Thanks also to Simon Garcia and Gabriel Merino for providing some of the figures.